
Введение
Повышенная проницаемость кишечного барьера является общим патофизиологическим механизмом для большой группы заболеваний, в первую очередь это характерно для воспалительных заболеваний кишечника (ВЗК) – болезни Крона (БК) и язвенного колита (ЯК).
Известно, что кишечный барьер представляет собой сложную многослойную систему, обеспечивающую, с одной стороны, защиту от проникновения антигенов, измененной толстокишечной микрофлоры и токсинов микроорганизмов, с другой – всасывание нутриентов и воды [1]. Физические компоненты этого барьера – слой слизи, эпителиальная выстилка и эндотелий сосудов. Медиаторы воспаления, антимикробные пептиды, иммуноглобулин А – химические составляющие кишечного барьера. При этом кишечная проницаемость определяется как функциональная характеристика кишечного барьера [2]. Оценка проницаемости по сывороточному уровню йогексола показала, что у 50% пациентов с БК и у 31% с ЯК она повышена и коррелирует с эндоскопической активностью заболевания [3]. Интересен установленный факт изменений параклеточной проницаемости у больных ВЗК с кишечными симптомами даже в отсутствие активности воспаления по данным эндоскопического исследования [4].
Кишечный эпителий состоит из монослоя различных подтипов кишечных эпителиальных клеток (IEC): энтероцитов, бокаловидных клеток, клеток Панета, эндокриноцитов, М-клеток, чашеобразных (cup cells) и пучковых клеток (tuft cells) [5]. Механическое соединение этих клеток и регуляция проницаемости для ионов и молекул обеспечиваются тремя типами соединительных комплексов: плотными контактами (TJ), адгезионными соединениями (AJ) и десмосомами (рис. 1).

Наиболее широко в литературе представлены характеристики TJ и AJ, объясняющие взаимосвязь между их структурными изменениями и повышением параклеточной проницаемости. Исследование биоптатов слизистой оболочки (СО) толстой кишки при ЯК и БК продемонстрировало ультраструктурные изменения в виде расширения апикальных соединений и увеличения межклеточного пространства [6, 7].
Плотные контакты (TJ)
Апикальный соединительный комплекс TJ представляет собой сложную сеть фибрилл и состоит из более 150 белков, включая клаудины, соединительные молекулы адгезии JAM-A (Junctional Adhesion Molecule-A), белки TAMP, содержащие домен Marvel (Tight Junction-Associated Marvel domain) [8]. Морфологические изменения, способствующие потере барьерной функции, при ЯК и БК имеют много общего: уменьшение количества горизонтально ориентированных нитей TJ и глубины сети TJ, разрывы нитей TJ, превышающие 25 нм [9].
Установлено, что основными белками, определяющими барьерные свойства и участвующими в регуляции параклеточного пути TJ, являются клаудины. В настоящее время идентифицировано 27 видов клаудинов, функционально включающих 2 группы – «порообразующие» и клаудины «запирающего» ряда [10]. ВЗК связаны со сниженной экспрессией «запирающих» клаудинов, в первую очередь клаудинов 1, 3, 4, 5, 7 и 8 [11]. Продукция различных цитокинов ответственна за подавление экспрессии герметизирующих клаудинов в воспаленной СО кишечника. Эти цитокины действуют на рецепторы энтероцитов, что активирует внутриклеточные сигнальные каскады и проводит к изменению активности факторов транскрипции в ядре. В условиях in vitro и in vivo было обнаружено, что фактор некроза опухоли α (TNF-α), интерферон γ (IFN-γ), интерлейкины (IL) 1β, 4, 6, и 13 снижают экспрессию клаудинов и увеличивают проницаемость эпителия [12].
В снижении экспрессии белков TJ и развитии ВЗК также играет роль дисбаланс кишечных микроРНК (microRNA, miRNA), которые представляют собой короткоцепочечные некодирующие молекулы РНК, посттранскрипционно контролирующие экспрессию большого разнообразия генов [13]. В частности, miR-29 уменьшает экспрессию клаудина-1, miR-223 – клаудина-8 [14, 15]. Из исследований в этом направлении представляем работу R.K. Felwick et al. (2020), в которой изучалось влияние microRNA23a на проницаемость эпителиального барьера (ЭБ) при БК. В качестве объекта для изучения использовались биоптаты сигмовидной кишки 16 пациентов с активной и 7 с неактивной формами заболевания, 10 здоровых добровольцев. Установлено, что microRNA23a сверхэкспрессируется при БК вне зависимости от активности процесса с подавлением продукции ингибитора TNFAIP3, что повышает чувствительность к TNF-α и увеличивает проницаемость толстой кишки [16].
В развитии повышенной параклеточной проницаемости при ВЗК играет роль и повышение экспрессии «поро-образующих» клаудинов, в частности клаудина-2. IL-13 и -6 являются мощным индуктором этого белка TJ в культивируемых эпителиальных клетках кишечника [17]. Кроме того, показано, что клаудин-2 вытесняет герметизирующий клаудин-4 из TJ, дополнительно ослабляя параклеточный барьер [18].
Cемейство белков TAMP включает окклюдин, трицеллюлин и белок MarvelD3 [19]. Это первые идентифицированные трансмембранные компоненты TJ, однако данные об их роли в параклеточной проницаемости противоречивы [20]. Так, ингибирование моноклональным антителом окклюдина в культивируемых клетках линии T84 ослабляло повторную сборку цепей TJ, а его подавление с помощью siRNA (small interfering RNA – малая интерферирующая РНК) в культуре клеток Сасо-2 задерживало развитие параклеточного барьера. Кроме этого сверхэкспрессия окклюдина в L-клетках не приводила к сборке TJ-подобных фибрилл, а подавление его экспрессии не сопровождалось очевидными нарушениями проницаемости ЭБ и развитием ВЗК [20]. По сведениям T. Kucharzik et al. (2001), у пациентов с ЯК и БК регистрировалось значительное снижение экспрессии окклюдина [21].
Соединительные молекулы адгезии (JAM) представляют собой гликопротеины, которые принадлежат к суперсемейству иммуноглобулинов (IgSF). В свою очередь семейство JAM состоит из белков: JAM-A, -B, -C, -4, JAM-подобного белка (JAM-L), рецептора Коксаки и аденовируса (CAR), CAR-подобного мембранного белка (CLMP) и молекулы избирательной адгезии эндотелиальных клеток (ESAM) [22]. В исследовании S. Vetrano et al. (2008) продемонстрирована роль JAM-A в контроле гомеостаза СО кишечника путем регулирования целостности и проницаемости ЭБ и установлено, что рецидивы БК и ЯК, а также экспериментальный колит сопровождаются снижением экспрессии JAM-A [23].
Мембранно-ассоциированные белки семейства гуанилаткиназы (MAGUK), окклюзионные соединения (известные как zonula occludens) ZO-1, -2, -3, а также цингулин, белки MAGI и комплекс полярности PAR3/PAR6 связываются с клаудинами, окклюдином, трицеллюлином, JAM-A и опосредуют их связь с актиновыми филаментами цитоскелета [24]. В исследованиях in vitro подавление ZO-2 и -3 не влияло на формирование TJs, в то время как подавление ZO-1 ослабляло сборку TJ и формирование ЭБ [25]. Это нашло подтверждение в недавней работе Y. Tan et al. (2019), где авторами показано, что пациенты с ЯК независимо от фазы заболевания имели повышенную экспрессию клаудина-2 и сниженную – окклюдина и ZO-1 по сравнению с контрольной группой здоровых лиц. Примечательно, что экспрессия ZO-1 была значительно выше у пациентов в процессе заживления СО толстой кишки. Авторы поддерживают точку зрения о том, что белки TJ играют важную роль в формировании эпителиального барьера и заживлении СО [26].
Адгезивные контакты (AJ)
Установлено, что основным трансмембранным белком в AJ является E-кадгерин. На цитоплазматической стороне AJ E-кадгерин связывается с p120-катенином, β-катенином и α-катенином, образуя комплекс, прикрепленный к кортикальным актиновым филаментам [27]. В клинических и экспериментальных исследованиях установлено нарушение строения AJ при ВЗК, что указывает на роль AJ в развитии дефектов кишечного барьера. Так, подавление экспрессии p120-катенина у мышей приводило к дефектам межклеточной адгезии, воспалению, прогрессирующей эрозии СО и терминальному кровотечению [28].
Интересные данные представлены в исследовании Ch. Zhang et al. (2015), в котором изучалась экспрессия белков E-кадгерина, p120ctn, β-катенина и ядерного фактора κB (NF-κB) в 23 образцах толстокишечных тканей после колэктомии пациентов с фульминантным ЯК и 17 образцах – без активного воспаления СО толстой кишки. В тканях толстой кишки у пациентов с ЯК были зарегистрированы разнонаправленные изменения в виде снижения экспрессии E-кадгерина, p120ctn и β-катенина и повышения уровня NF-κB, что свидетельствовало о нарушении кишечного ЭБ [29].
Нарушение транспорта белков AJ и TJ
Снижение экспрессии белков AJ и TJ – не единственный механизм, лежащий в основе развития повышенной проницаемости ЭБ, т.к. эти белки перераспределяются из межклеточных контактов в компартменты [30]. Нарушение строения AJ и TJ может быть вызвано усилением эндоцитоза либо уменьшением экзоцитоза соединительных белков и вносить вклад в разрушение ЭБ, однако механизмы, регулирующие интернализацию белков в норме и при ВЗК, остаются плохо изученными. В подавляющем большинстве исследований в этой области использовались модельные системы in vitro [31, 33].
Установлено, что в ответ на воздействие IFN-γ происходит интернализация белков TJ за счет макропиноцитоза в эндосомы. Эндотелиальные клетки, обработанные IFN-β, блокировали индуцированный IFN-γ эндоцитоз и поддерживали целостность барьера. Обращает внимание, что при биопсии СО толстой кишки пациентов с активным ЯК обнаруживаются интернализованные субапикальные пузырьки, подобные тем, что обнаруживаются в культивируемых клетках Т84, обработанных IFN-γ [32].
В исследовании D. Smyth et al. (2012) на модели T84 обнаружено, что IFN-γ-индуцированная интернализация E-кадгерина требует его убиквитинирования, которое опосредуется тирозинкиназой Src и убиквитинлигазой Hakai (E3 ubiquitin ligase). Исследования иммунопреципитации продемонстрировали снижение E-кадгерина в мембранной фракции и соответствующее увеличение цитозольного E-кадгерина и связанных с ним p120-катенина и β-катенина [33].
Экзоцитоз белков AJ и TJ представляет собой их транспорт из клетки к плазматической мембране. Он включает доставку из эндоплазматического ретикулума и комплекса Гольджи не только вновь синтезированных белков, но и ранее интернализованных молекулярных компонентов AJs и TJs [34].
В исследовании J.A. Rodríguez-Feo et al. (2015) изучалось влияние структурного белка канальцев эндоплазматического ретикулума ретикулона-4B (RTN-4B/NOGO-B), на барьерную функцию кишечника. Подавление экспрессии ретикулона-4B в монослоях кишечных эпителиальных клеток приводило к снижению экспрессии E-кадгерина, α-катенина и окклюдина. Анализ толстокишечных биоптатов у пациентов с ВЗК показал значительное снижение экспрессии RTN-4B/NOGO-B по сравнению с тканями СО здоровых добровольцев [35].
Значение переноса белков в проницаемости кишечного барьера продемонстрировано исследованиями регулятора слияния везикул – белка прикрепления NSF-α (αSNAP). Этот белок контролирует перенос везикул между эндоплазматическим ретикулумом и аппаратом Гольджи [36]. Подавление αSNAP в клетках SK-CO15 сопровождалось нарушением целостности барьера, разрушением AJ и TJ и приводило избирательному снижению экспрессии E-кадгерина и p120-катенина [37].
Нарушение цитоскелета
Апикальная область эпителиальных клеток богата актиновыми филаментами (F-актин) и немышечным миозином II (NMII), собранных в несколько структур. Связь белков AJ и TJ с актиновым цитоскелетом является важной составляющей целостности кишечного ЭБ. По данным литературы, белки TJ, по-видимому, связаны со стабильными филаментами, состоящими из γ-цитоплазматического актина (γ-CYA), тогда как AJ соединяются с более динамичными филаментами, β-цитоплазматической изоформой актина (β-CYA) [38].
Нарушение актинового цитоскелета наблюдалось в модельных монослоях кишечных эпителиальных клеток, которые подвергались воздействию различных медиаторов воспаления. Анализ тканей СО у пациентов с ВЗК показал, что ее воспаление вызывает значительные изменения в экспрессии самого актина и ряда актин-связывающих белков [39].
Хорошо известно, что актиновые филаменты обеспечиваются сложной регуляцией с участием большого количества вспомогательных, сигнальных и моторных белков. Оборот F-актина регулируется различными актин-связывающими белками (ABP), ответственными за процессы полимеризации или деполимеризации. В свою очередь полимеризация координируется комплексом Arp 2/3 (actin related protein) и форминами, деполимеризация – членами семейства актин-деполимеризующих факторов (ADF)/кофилинов [40]. Уменьшение экспрессии компонентов комплекса Arp 2/3 в кишечнике Caenorhabditis elegans привело к снижению уровня апикального F-актина и некоторых апикально связанных белков [41].
Однако в исследовании K. Zhouу et al. (2015) у мышей с отсутствием ArpC3 (компонента комплекса Arp 2/3) значительного снижения содержания кортикального F-актина не выявлено. Были обнаружены дефекты эндолизосомной системы и ограничение всасывания питательных веществ [42]. Ингибирование ADF или кофилина-1 посредством РНК-интерференции увеличивало параклеточную проницаемость культивированных клеток HT-29 вследствие задержки сборки филаментозного актинового пояса. Кроме того, ADF-нулевые мыши продемонстрировали повышенную кишечную проницаемость при колите, индуцированном декстрансульфатом натрия [43].
Эпителиальные клетки экспрессируют немышечный миозин II, состоящий из двух тяжелых цепей: двух основных (MLC) и двух регуляторных легких цепей (RMLC). N.G. Naydenov et al. (2016) провели исследование, в котором изучалось влияние ингибирования тяжелой цепи миозина NM IIA на структуру и функцию кишечного барьера у мышей, а также развитие у них экспериментального колита. Была отмечена повышенная кишечная проницаемость и измененная экспрессия нескольких белков AJ/TJ.
Следует отметить, что в случаях отсутствия у мышей колита при морфологическом исследовании регистрировались признаки слабого воспаления, инфильтрация нейтрофилами СО толстой кишки, а также повышенная экспрессия цитокинов.
Повреждение СО при DSS-инду-цированном колите было более выражено по сравнению с контролем [44].
Исследования in vitro показывают, что дисфункция эпителиального барьера может быть опосредована повышенной экспрессией киназы MLCK и последующим фосфорилированием RMLC [45]. Для подтверждения этого наблюдения S.A. Blair et al. (2006) проанализировали указанные процессы в образцах биоптатов СО толстой кишки пациентов с ВЗК с помощью количественной иммунофлуоресцентной микроскопии. Авторы отмечают, что экспрессия MLCK увеличивается при ВЗК и коррелирует с активностью заболевания, что свидетельствуют о ее роли в состоятельности кишечного барьера [46].
Молекулярная архитектура и динамика цитоскелета регулируются различными сигнальными событиями. Установлено, что важными сигнальными молекулами являются члены суперсемейства Ras малых ГТФ-аз (GTPases). Суперсемейство Ras состоит из 6 подсемейств, а именно Ras, Rho, Arf, Ran, Rab, Rit. Некоторые ГТФазы оказывают синергетическое действие на кишечный ЭБ, в то время как другие обладают довольно уникальными функциями [47].
В литературе можно найти достаточное количество исследований, описывающих важную роль Rho GTPases в формировании эпителиального барьера и их способность контролировать динамику актина. Наиболее подробно описаны члены семейства Rho: RhoA, RhoB, Rac1 и Cdc42. Например, ингибирование эффектора RhoA, Rho-kinase (ROCK) в культивированных клетках T84 нарушает строение актомиозинового комплекса и ведет к увеличению проницаемости кишечного барьера [48]. J. Melendez et al. (2013) установили, что тотальный дефицит Cdc42 у мышей приводит к повышению проницаемости ЭБ и индуцирует нарушение гомеостаза СО кишечника [49]. Y. Yang et al. (2013) пришли к выводу, согласно которому miR-21 индуцирует деградацию мРНК RhoB, приводя к истощению белка RhoB, дисфункции TJ и повышению проницаемости, подтверждая важную роль как miR-21, так и RhoB в гомеостазе ЭБ у пациентов с ЯК [50].
Заключение
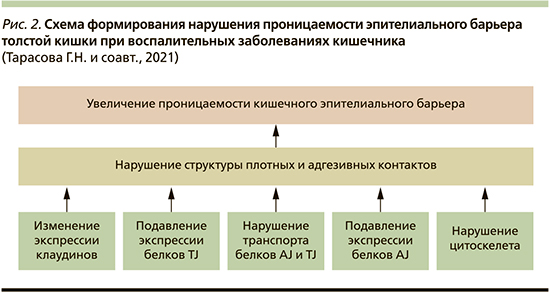
Нарушение барьерной функции кишечника – одно из ключевых событий в патогенезе ЯК и БК (рис. 2). Однако до сих пор неизвестно, является ли нарушенный барьер следствием продолжающегося воспаления, или это независимый процесс, связанный с патофизиологией ВЗК. Механизмы, регулирующие кишечный эпителиальный гомеостаз, сложные и многоступенчатые. Расширение нашего понимания о механизмах воспалительно-зависимых изменений в проницаемости эпителия даст новые идеи для разработки терапевтических средств с целью улучшения заживления СО толстой кишки при ВЗК.
Авторы подтверждают отсутствие конфликта интересов, о котором необходимо сообщить.


