
Синдром Луи-Бар (D. Louis-Bar) впервые был описан в 1941 г. во Франции. Частота встречаемости составляет 1 случай на 40 тыс. новорожденных. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к т.н. факоматозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа–Вебера, туберозный склероз и др. В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т.е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей [1]. Наиболее часто синдром Луи-Бар начинает проявляться в возрасте от 5 месяцев до 3 лет. При этом отмечается появление мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание, качание туловища и головы во время движения. Характерна мозжечковая дизартрия, характеризующаяся невнятной скандированной речью. Отмечаются мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие. Имеется склонность к инфекционным заболеваниям: частые острые респираторно-вирусные инфекции, пневмонии, имеющие затяжное течение и плохо поддающиеся лечению. Нередки опухоли яичников, желудка, кожи.
 Телеангиэктазии появляются в возрасте от 3 до 6 лет и первоначально возникают на конъюнктиве глазного яблока, затем на коже век, носа, лица и шеи, локтевых и коленных сгибах, предплечьях, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба, но наиболее выражены в тех местах кожного покрова, которые подвергаются воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». Кожа при этом теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии. Из других кожных проявлений встречаются веснушки, пятна цвета «кофе с молоком», участки обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Могут наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне, или проявления псориаза [2].
Телеангиэктазии появляются в возрасте от 3 до 6 лет и первоначально возникают на конъюнктиве глазного яблока, затем на коже век, носа, лица и шеи, локтевых и коленных сгибах, предплечьях, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба, но наиболее выражены в тех местах кожного покрова, которые подвергаются воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». Кожа при этом теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии. Из других кожных проявлений встречаются веснушки, пятна цвета «кофе с молоком», участки обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Могут наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне, или проявления псориаза [2].
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у трети пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов (Ig) крови, которое выявляет значительное снижение IgA и IgЕ, в 10–12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться ультразвуковое исследование тимуса, при котором может быть диагностирована аплазия или гипоплазия тимуса; магнитно-резонансная томография головного мозга с целью выявления атрофии мозжечка, расширения IV желудочка; фарингоскопия, риноскопия. Также выполняется рентгенография легких для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений [3, 4]. Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю–Ослера, атаксией Пьера–Мари, синдромом Гиппеля–Линдау и др.
Выявление синдрома Луи-Бар требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи–Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
 К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокорригирующая терапия препаратами тимуса (Т-активин) и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокорригирующая терапия препаратами тимуса (Т-активин) и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
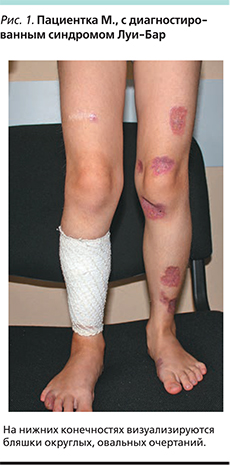
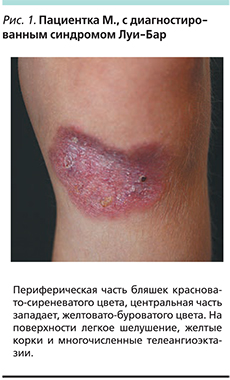
Под нашим наблюдением находилась девочка М. 5 лет, у которой в год и 3 месяца обнаружилась атаксия. Приблизительно в двухлетнем возрасте появились расширенный сосуд на правой конъюнктиве, папулезная сыпь на нижних конечностях. В 2012 г. на основании клинической картины, наличия первичного иммунодефицита (снижение уровней IgA, -G), ДНК-диагностики (наличие мутированного гена АТМ) был поставлен диагноз «атаксия–телеангиэктазия». При гистологическом исследовании биоптатов кожи был обнаружен продуктивный гранулематозный дерматит, вероятно инфекционной природы, который не был подтвержден ни клинически, ни лабораторно. Девочка получала заместительную терапию препаратами иммуноглобулинов, биологическими агентами, симптоматическими средствами, местно кремом, содержащим такролимус. В 2014 г. проведено повторное гистологическое исследование кожных высыпаний, по результатам которого было получено заключение: саркоидозный дерматит. Проведена рентгеновская компьютерная томография органов брюшной и грудной полостей, которая не выявила патологии. С 2015 г. отмечено ухудшение кожного процесса с увеличением размера высыпаний и появления в их центре изъязвлений без болезненных ощущений и лихорадки. Проведено очередное гистологическое исследование на кафедре анатомии МГМУ. И.М. Сеченова, которое выявило множественные сливающиеся эпителиоидноклеточные и гигантоклеточные гранулемы со скоплением макрофагов, отдельных гигантских многоядерных клеток типа Пирогова–Лангханса, а также лимфоцитов. Гранулемы локализуются в дерме и окружены валом воспалительного лимфогистиоцитарного инфильтрата. Часть гранулем имеют в центре казеозный некроз. Было получено заключение о необходимости проведения дифференциального диагноза между саркоидозом, микобактериозом и туберкулезом. При осмотре: состояние тяжелое по основному заболеванию. Жалобы на шаткость походки, кожные высыпания. Местный статус: на нижних конечностях, ягодицах имеются бляшки округлых овальных очертаний диаметром 5–8 см. Периферическая часть бляшек красновато-сиреневатого цвета, центральная часть западает желтовато-буроватого цвета. На поверхности легкое шелушение, желтые корки и многочисленные телеангиоэктазии (рис. 1, 2).
С учетом клинических проявлений кожного процесса, гистологических исследований, отсутствия данных за саркоидоз и туберкулез можно думать о липоидном некробиозе. Причем отсутствие липоидных отложений в биоптате говорит в пользу гранулематоза Мишера–Ледера, который, впрочем, также, по мнению ряда гистологов, представляет собой липоидный некробиоз [5]. Данный случай представляет особый интерес, т.к. в доступной литературе мы не нашли описания подобных кожных проявлений при этом редком генетическом заболевании. Тем более что врачи других специальностей не знакомы с редкой кожной патологией, что вызвало неправильную трактовку гистологического исследования. Фибриноидная дегенерация коллагена, имеющая место при липоидном некробиозе, скорее всего была принята за казеозный некроз, что и привело к заключению о микробной этиологии высыпаний, хотя тщательное обследование больной на туберкулез дало отрицательный результат.


