
Нефрокальциноз является одной из актуальных проблем в детской нефрологии. В последнее время отмечается увеличение диагностирования нефролитиаза среди детского населения, что, вероятно, связано с широким внедрением в диагностику скрининговых методов обследования и новейших визуализирующих технологий.
Первичная гипероксалурия – это группа аутосомно-рецессивных заболеваний, характеризующаяся гиперпродукцией оксалата вследствие дефицита ферментов обмена глиоксиловой кислоты, что вызывает резкое усиление внепочечного биосинтеза оксалатов [6, 12]. На сегодняшний день идентифицировано три формы первичной гипероксалурии, среди которых 1-й тип (OMIM 259900) является наиболее распространенным, а также самым тяжелым [9, 12].
Оксалоз 1-го типа составляет 80% от всех случаев первичной гипероксалурии. Заболевание вызвано дефицитом или полным отсутствием пероксисомного печеночного фермента L-аланина: глиоксилата аминотрансферазы (AGT). Распространенность первичной гипероксалурии составляет от 1 до 3,4 на 1 млн населения в Северной Америке и Европе, достоверных данных по Российской Федерации в настоящее время не имеется [4, 5, 13].
Гипероксалурии 2-го (OMIM 260000) и 3-го типов (OMIM 613616), соответственно вызваны дефицитом глиоксилат редуктазы/гидроксипируват-редуктазы (GRHPR) и 4-гидрокси-2-оксо-глутарат-альдолазы и составляют приблизительно 10% всех первичных гипероксалурий [2, 4, 7]. Генетически обусловленное нарушение метаболизма глиоксиловой кислоты приводит к усиленному биосинтезу оксалата и повышению почечной экскреции солей щавелевой кислоты. Избыток оксалата выводится с мочой, где он выпадает в осадок в виде кристаллов кальция оксалата моногидрата (вевеллит). Оксалатно-кальциевые кристаллы откладываются в почечной паренхиме и других органах и тканях с развитием рецидивирующего нефролитиаза и прогрессирующим снижением скорости клубочковой фильтрации (СКФ) [3, 6].
Клиническая картина значительно варьируется: от рецидивирующего нефролитиаза и почечной недостаточности в раннем возрасте до случайно выявленных форм в зрелом возрасте. От 20 до 35% случаев заболевания диагностируются на стадии терминальной хронической почечной недостаточности. Почечная недостаточность обычно возникает у пациентов с первичной гипероксалурией 1-го или 2-го типа, в редких случаях у больных с 3-м типом, для которого характерно наименее тяжелое течение [1, 9]. Выделяют инфантильную (4–12 месяцев), ювенильную (после 1 года) и поздно манифестирующую формы (после 25 лет). В 15% случаев заболевание диагностируется в возрасте до 1 года и характеризуется ранним развитием терминальной хронической почечной недостаточности и системным паренхиматозным оксалозом. Ювенильные формы заболевания чаще выявляются до 5-летнего возраста и характеризуются более медленным темпом снижения клубочковой фильтрации. Следует отметить, что поздно манифестирующие случаи первичной гипероксалурии проявляются скудной клинической симптоматикой с редким отхождением конкрементов и медленным развитием терминальной почечной недостаточности [5, 7].
В целом большинство пациентов с первичной гипероксалурией в конечном счете нуждаются в заместительной почечной терапии с последующей трансплантацией. При снижении скорости клубочковой фильтрации ниже 40 мл/мин/1,73 м2 почки уже не в состоянии устранять избыток оксалата. В результате этого плазменные уровни оксалата возрастают и возникает системный оксалоз [7, 9]. Это осложнение может поражать практически любой орган, особенно часто кости, кровеносные сосуды, сердце и сетчатку. Внепочечные депозиты оксалата кальция варьируются по своему количеству и локализации. В основном кристаллы встречаются в головном мозге, костях, хрящах, часто в стенках сосудов, легких, лимфатических узлах, щитовидной, паращитовидной и вилочковой железах, селезенке, поджелудочной железе и надпочечниках [7, 12].
Диагностика оксалоза основывается на клинической картине и лабораторных исследованиях, включающих в первую очередь определение уровней суточной экскреции оксалатов в пересчете на креатинин мочи. У здоровых детей суточная экскреция оксалата не превышает 45 мг/сут или 0,5 ммоль/сут на 1,73 м2 поверхности тела. У больных с гипероксалурией этот показатель выше нормы в 2–4 раза. При прогрессировании почечной недостаточности экскреция оксалата резко снижается, однако нарастает его плазменная концентрация [4, 6].
Дифференцировать два типа первичной гипероксалурии можно по результатам определения органических кислот: для 1-го типа характерна экскреция гликолевой и глиоксиловой кислот, для 2-го – L-глицериновой кислоты. При митохондриальной дислокации фермента АGT (аланин глиоксилат аминотрансферазы), уровень гликолевой кислоты может быть в пределах нормы [4, 8].
Наличие дефицита фермента аланин-глиоксилат аминотрансферазы может быть также подтверждено биопсией печени, особенно в случаях, когда экскреция гликолата не нарушена. Оценка печеночных биоптатов включает количественную оценку ферментативной активности, иммуноблоттинг и иммуноэлектрофорез белка с целью определения каталитической активности фермента. Следует учитывать, что при дислокации фермента из пероксисом в митохондрии печени активность глиоксилат аминотрансферазы остается нормальной [10, 11].
На современном этапе наиболее информативным диагностическим критерием является молекулярно-генетическое исследование с выявлением мутации в генах AGXT, GRHPR, HOGA1, ответственных за обмен оксалатов. Ген AGXT состоит из 11 экзонов и локализован на хромосоме 2q37.3. Наиболее часто мутации затрагивают 1-й, 2, 4 и 10-й экзоны гена [5, 12, 14]. Описано более 170 мутаций, которые в большинстве случаев приводят к значительному уменьшению или полной потере ферментативной активности. Большинство из них – одиночные нуклеотидные изменения с c.508G> A (p.Gly170Arg), происходящие в 30% случаев мутантных аллелей среди всех пациентов [15].
Лечебные мероприятия при оксалозе направлены на снижение синтеза и кристаллизации оксалата кальция, а также предупреждение образования оксалатно-кальциевых камней. При определенных мутациях в гене AGXT (p.Gly170Arg, p.Phe152Ile) доказан положительный эффект от использования высоких доз (10–30 мг/кг/24 ч) пиридоксина. В некоторых случаях в сочетании с повышенной гидратацией терапия пиридоксином приводит к нормализации экскреции оксалатов мочи и может оказать влияние на исход трансплантации [16]. Ответом на терапию пиридоксином считается снижение экскреции оксалатов минимум на 30% от исходного уровня [10,12].
В качестве примера успешного использования такой терапии представляем клинический случай первичной гипероксалурии 1-го типа.
Мальчик Саша Л., возраст 11 месяцев, от неродственного брака, из семьи с неуточненным анамнезом по заболеваниям почек. Ребенок от первой беременности женщины 40 лет, протекавшей на фоне обострения пиелонефрита и гестационной артериальной гипертензии во втором и третьем триместрах, отмечена угроза прерывания беременности на всем протяжении гестации. В связи со слабостью родовой деятельности и нарастающей внутриутробной гипоксией плода родоразрешение проводилось путем кесарева сечения. Отмечена мекониальная окраска околоплодных вод. Родился с нормальными антропометрическими показателями: массой тела – 3290 г, рост – 53 см, оценка по шкале Апгар составила 8/8 баллов. Период адаптации протекал с токсической эритемой, неонатальной желтухой. Раннее психомоторное развитие соответствовало возрасту. Отмечена задержка физического развития, плохая прибавка массы тела.
При проведении УЗИ скрининга органов брюшной полости в возрасте 1 месяца жизни были выявлены гиперэхогенные образования в пирамидках почек до 7 мм, расцененные как нефрокальциноз. По данным анализов мочи, выявлялась рецидивирующая лейкоцитурия, микропротеинурия, микрогематурия. Ребенок наблюдался амбулаторно с диагнозом: нефрокальциноз неясного генеза. Вторичный хронический пиелонефрит, рецидивирующее течение. По месту жительства было рекомендовано проведение генетического исследования с целью исключения первичной гипероксалурии. Получал антибактериальную, мембраностабилизирующую терапию. При морфологическом обследовании мочевого осадка неоднократно выявлялась недифференцируемая клеточная пролиферация (атипичная почечная эпителиурия и лейкоцитурия), в связи с чем в возрасте 9 мес госпитализирован в хирургическое отделение по месту жительства с подозрением на новообразование почек и мочевого пузыря. Мальчик консультирован фтизиатром и онкологом. Убедительных данных за туберкулез мочевой системы получено не было. По данным заключения онколога, характер течения заболевания требовал тщательного наблюдения в динамике.
В возрасте 11 месяцев ребенок впервые поступил в педиатрическое отделение Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева РНИМУ им. Н.И. Пирогова Минздрава России с целью уточнения диагноза и определения дальнейшей тактики ведения. При поступлении состояние ребенка средней тяжести. При осмотре были отмечены следующие фенотипические особенности ребенка: низкие показатели физического развития – масса и рост соответствовали 3-й перцентили (рост – 69 см, масса – 7600 г), наличие малых аномалий развития: голова гидроцефальной формы, оттопыренные и низко посаженные ушные раковины, кожная форма синдактилии 2-го и 3-го пальцев стоп. Кожные покровы чистые, бледные, диффузная мышечная гипотония. Отеков нет. Тоны сердца отчетливые, ритмичные, систолический шум на верхушке. В легких пуэрильное дыхание, проводится во все отделы, хрипов нет. Живот мягкий, умеренно болезненный при пальпации во всех отделах. Гепатоспленомегалии не отмечено. Диурез в норме. Моча мутная. Стул регулярный.
По данным клинического анализа крови выявлена гипохромная анемия 1-й степени (уровень гемоглобина – 97 г/л). Мочевой синдром представлен абактериальной лейкоцитурией до 118 клеток в поле зрения, микрогематурией, микропротеинурией. Кислотно-основной состав крови в пределах возрастных значений. В биохимическом анализе крови отмечено повышение ионизированного кальция 1,37 ммоль/л (норма – 1,13–1,32 ммоль/л) при нормальном уровне общего кальция 2,46 ммоль/л (норма до 2,60 ммоль/л), уровень калия, фосфора и магния в пределах нормальных значений. Отмечено снижение уровня сывороточного железа до 3,4 мкмоль/л (норма – 6,6–28,3 мкмоль/л). Креатинин – 43 мкмоль/л, снижение СКФ по формуле Шварца до 47,4 мл/мин/1,73 м2. Уровень паратгормона находился в пределах возрастных значений 16,9 (при норме 16–62 пг/мл).
Суточная мочевая экскреция оксалатов была повышенной (Ox/Cr=0,43, при норме <0,1 ммоль/ммоль), тогда как экскреция кальция (Ca/Cr=0,34, при норме до 2,2 ммоль/ммоль), фосфора (P/Cr=2,85, при норме до 18 ммоль/ ммоль), уратов (Ur/Cr=0,85, при норме – 0,7–1,5 ммоль/ммоль) в пределах нормы; качественная реакция на цистин отрицательная. При исследовании экскреции органических кислот выявлено повышение уровня гликолевой кислоты (гликолевая кислота/Cr=214,3 при норме – 28–126 ммоль/ ммоль). Нарушена концентрационная функция почек в пробе Зимницкого – удельный вес мочи колебался в пределах 1002–1006.
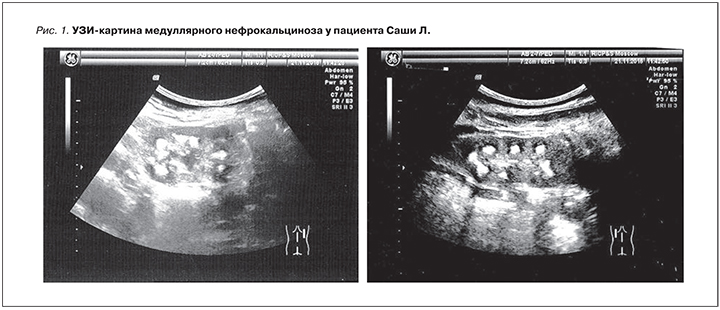
Морфология мочевого осадка: нейтрофилы – 69%, лимфоциты – 18%, эозинофилы – 13%. Обнаружены клетки эпителия с признаками атипии в большом количестве. По данным УЗИ почек выявлены утолщение и диффузные изменения почечной паренхимы, симптом «гиперэхогенных пирамидок», эхогенность коркового слоя значительно повышена, сравнима с эхогенностью печени. По периферии пирамидок эхогенность не изменена, что соответствует нефрокальцинозу второй стадии. Корковый слой истончен. Почки несколько увеличены в размерах (правая 5,3×3,4×2,8 см; объем – 26,6 см3, левая 5,9×3,7×2,6 см3; объем – 29,2 см3, соотношение объема почек и массы тела: 0,73% норма (0,4– 0,7)% (рис. 1).
При допплерографическом исследовании сосудов почек выявлены пониженная систолическая скорость кровотока на уровне почечных, сегментарных, долевых артерий обеих почек и повышенные показатели периферического сопротивления на уровне почечных, сегментарных, долевых артерий обеих почек.
Мальчик обследован в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра Москвы в возрасте 10 месяцев (12.10.2016) с целью исключения первичной гипероксалурии. Методом прямого секвенирования были проанализированы 1-й, 2, 4, 8–10-й экзоны гена AGXT. Выявлены замены Pro11Leu (СМ910014) в гомозиготном состоянии и замена lle340Met (CM910018) в гомозиготном состоянии, которые являются однонуклеотидными полиморфизмами. В 4-м экзоне выявлена мутация Gly170Arg (CM910016), приводящая к замене с.508G>A в гомозиготном состоянии. Было рекомендовано исследовать ДНК родителей.
Таким образом, ребенку из семьи с неуточненным анамнезом по патологии почек, с признаками нефрокальциноза по результатам УЗИ почек, рецидивирующей абактериальной лейкоцитурией, микропротеинурией и микрогематурией, с молекулярно-генетически подтвержденной мутацией в гене AGXT (в 4-м экзоне выявлена гомозиготная мутация GLy170Arg, приводящая к замене с.508G>A) и повышением уровня гликолевой кислоты был установлен диагноз «первичная гипероксалурия 1-го типа».
С целью выявления носительства мутации р.GLy170Arg(СМ910016) были обследованы родители мальчика. По данным лаборатории селективного скрининга Медико-генетического научного центра Москвы, было установлено, что у матери и отца-пробанда мутация р.GLy170Arg в гетерозиготном состоянии (носители). В связи с выявленными изменениями в уроцитограмме в виде недифференцируемой клеточной пролиферации ребенок был переведен в отделение онкоурологии Российского научного центра рентгенорадиологии с целью исключения уротелиальной опухоли.
Ребенку была проведена магнитно-резонансная томография органов малого таза с внутривенным контрастированием. Очаговых и объемных изменений органов малого таза не выявлено. Выполнен забор мочи на цитологическое исследование из правого и левого мочеточников раздельно. В материале осадка – эритроциты, нейтрофильные лейкоциты, клетки уротелия с реактивными изменениями (категория II, согласно парижской системе оценки цитопатологии уринарного тракта – негативные по уротелиальной карциноме высокой степени злокачественности). Также была проведена уретроцистоскопия с последующей биопсией мочевого пузыря. При гистологическом исследовании материала отмечен рост фиброзной ткани с сосудами в подслизистой оболочке, опухолевые клетки не обнаружены. Таким образом, данных за злокачественное образование мочевого пузыря выявлено не было. Рекомендовано динамическое наблюдение через 6 месяцев с повторным проведением цитологического исследования мочи.
При последующем катамнестическом наблюдении за ребенком в течение года (2016–2017) на фоне проведения патогенетически обоснованной терапии высокими дозами пиридоксина (15 мг/кг/ сут), антикристаллобразующими препаратами (цитратная смесь – Блемарен под контролем рН мочи в диапазоне 6,5–6,8, препараты магния – магне В6), у ребенка отмечена нормализация оксалатно-креатининового индекса (Ox/Cr=0,13 ммоль/ммоль при норме <0,16 ммоль/ммоль). При повторных исследованиях мочевого осадка атипичных клеток обнаружено не было.
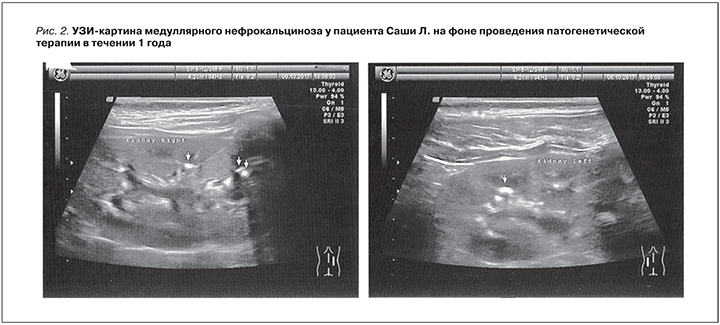
Наблюдалось уменьшение размеров гиперэхогенных включений при проведении контрольного УЗИ почек (рис. 2). Повысилась фильтрационная функция почек (СКФ по Шварцу – 79 мл/мин/1,73 м2).
Планируется продолжение патогенетической терапии пиридоксином и обязательное обсуждение с трансплантологами дальнейшей тактики ведения ребенка с целью определения вида и срока проведения трансплантации и подготовки ребенка к будущей операции.
Приведенное клиническое наблюдение демонстрирует необходимость индивидуального подхода к диагностике и прогнозированию течения при первичной гипероксалурии с учетом молекулярно-генетического варианта патологии, что важно для определения стратегии ведения пациента.


