
Миома матки (ММ, также известная как лейомиома) является наиболее распространенной доброкачественной опухолью женской репродуктивной системы [1, 2]. ММ представляет собой доброкачественную моноклональную опухоль, происходящую из гладкомышечных клеток шейки или тела матки [3, 4]. К моменту наступления менопаузы более 70% женщин имеют ММ. Она диагностируется у 20–40% женщин репродуктивного возраста [4] с частотой в течение жизни от 30 до 70% [5]. После менопаузы ММ часто регрессируют. Аналогичные показатели распространенности ММ характерны и для женщин в Российской Федерации [6]. В то время как у многих женщин ММ протекает бессимптомно, около четверти из них имеют ее клинические проявления [7] и потенциальное нарушение репродуктивной функции, а также другие признаки общего влияния ММ на качество жизни, связанное со здоровьем. При наступлении беременности у пациенток с ММ ее течение в 10–40% сопровождается осложнениями, а выкидыши у женщин с симптомной ММ встречаются в два раза чаще [8].
ММ является основной причиной гинекологической заболеваемости, одной из наиболее распространенных причин гинекологических госпитализаций и наиболее частой причиной гистерэктомии в США [9]. В Российской Федерации ММ является причиной гистерэктомии в 50–70% случаев [10]. Прогнозируется, что годовая стоимость лечения этого состояния в США превысит 34 млрд долларов, что выше, чем стоимость лечения рака груди и толстой кишки, вместе взятых [11]. Кроме того, бесплодие и проблемы с беременностью, связанные с ММ, могут увеличить общие затраты и заболеваемость [9]. ММ может влиять на повседневную деятельность, межличностные отношения и производительность труда [12]. Симптомные миомы негативно влияют на качество жизни женщин, которое ухудшается по мере увеличения количества и выраженности симптомов.
ММ представляет собой сложное заболевание, вызванное сочетанием нескольких демографических, диетических и гормональных факторов риска [9, 13–15], а также биологических, эпигенетических и генетических причин [16, 17]; генетический компонент составляет от 40 до 50% [18]. В последние годы многие ученые исследуют генетические основы образования, развития и прогрессирования ММ.
В настоящее время нигде в мире не используются данные о влиянии генотипов однонуклеотидного полиморфизма на риск развития или рецидива миомы, хотя прогнозирование такого риска может помочь в принятии клинических решений. Низкие отношения шансов для всех известных полиморфных локусов косвенно свидетельствуют о том, что генетический риск развития миомы обычно определяется совокупностью нескольких факторов [15]. Учитывая это, разумным подходом к разработке диагностических систем является использование многофакторных методов оценки генетического риска с использованием генотипирования одновременно по нескольким локусам. Однако единственной известной работой, использующей этот принцип, остается опубликованное в 2012 г. исследование Bongadgi N.S. et al. [19]. Описаны генетические детерминанты, каждая из которых сама по себе несет высокий риск заболевания [20, 21], но такие маркеры крайне редко встречаются в популяции и, как следствие, вносят незначительный вклад в общую заболеваемость. Кроме того, известные детерминанты этого типа ограничены отдельными этно-территориальными группами, что не позволяет использовать их в других популяциях.
Полногеномный анализ ассоциаций (ПГАА, Genome-Wide Association Studies – GWAS) используется для изучения роли генетических маркеров в возникновении и прогрессировании различных многофакторных заболеваний, в том числе ММ [22]. В настоящее время генетические основы ММ активно изучаются различными научными коллективами с применением ПГАА. В то же время опубликованные данные иногда противоречат друг другу и имеют низкую воспроизводимость в различных группах населения.
Следует отметить, что население России характеризуется значительным этно-территориальным разнообразием, что определяет выраженные различия в генетической структуре разных народов России, и это может оказать существенное влияние на результаты исследований генов-кандидатов, ассоциированных с ММ. Этот фактор необходимо учитывать при планировании и проведении генетико-эпидемиологических исследований ММ в российских популяциях.
Целью данного обзора было проведение анализа опубликованных полногеномных исследований ММ с целью выявления ПГАА-значимых полиморфных локусов, ассоциированных с ММ.
Материалы и методы
Поиск публикаций осуществлялся в электронных базах данных PubMed, PubMed Central, eLibrary и в каталоге GWAS (ПГАА) за период с 2011 г. по настоящее время. Для поиска использовались следующие ключевые слова: «uterine leiomyomas», «fibroids», «GWAS studies», «candidate genes».
Результаты и обсуждение
На момент написания (июль 2022 г.) каталог полногеномных исследований Национального института изучения генома человека (https://www.ebi.ac.uk/gwas/) включает результаты восьми исследований, в которых изучалась предрасположенность к ММ.
Первое ПГАА-исследование ММ провели Cha P.C. et al. на выборке японских женщин в 2011 г. Всего было протестировано 457 044 однонуклеотидных полиморфизма (SNP) на их связь с ММ в 1612 клинически подтвержденных случаях, в которых были выявлены тяжелые симптомы ММ, включая дисменорею или гиперменорею, или в анамнезе имелись гистерэктомия, миомэктомия или эмболизация маточных артерий из японского проекта BioBank и 1428 контролей без ММ в анамнезе. Затем эта связь была воспроизведена в независимой японской когорте из 3466 случаев и 3245 контролей без ММ в анамнезе. Было показано, что три локуса на хромосомах 10q24.33, 22q13.1 и 11p15.5 имеют значимые полногеномные корреляции с ММ. SNP, демонстрирующие наиболее значимую ассоциацию в комбинированном анализе в каждом из этих локусов, – rs7913069 (p=8,65×10-14, отношение шансов (ОШ)=1,47), rs12484776 (p=2,79×10-12, ОШ=1,23) и rs2280543 (p=3,82×10-12, ОШ=1,39) соответственно (табл. 1) [23].
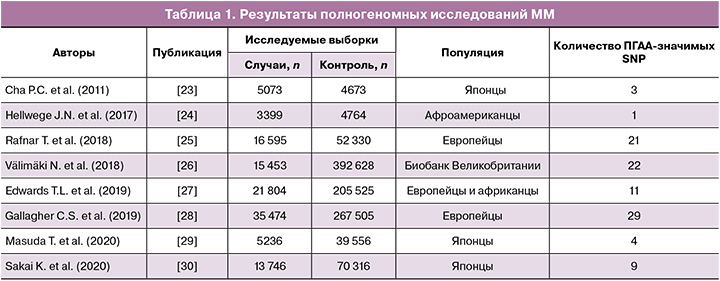
Hellwege J.N. et al. опубликовали первое многоэтапное полногеномное исследование ассоциаций риска ММ у афроамериканок в 2017 г., в котором было изучено 675 206 SNP. Ассоциации с риском миомы были смоделированы с использованием логистической регрессии с поправкой на основные компоненты, а результаты были подвергнуты метаанализу. В нем была выявлена значимая ассоциация между 3399 случаями и 4764 контролями (были подтверждены с помощью визуализации органов малого таза, генотипированы и причислены к 1000 геномам) при rs739187 (частота аллеля риска=0,27) в CYTH4 (ОШ (95% доверительный интервал)=1,23 (1,16–1,30), p=7,82×10-9).
Этот первый многоэтапный ПГАА миомы среди афроамериканок обнаружил новые локусы риска миомы в пределах CYTH4 с вероятным биологическим значением для миомы [24].
Rafnar T. et al. в 2018 г. опубликовали первый ПГАА ММ в европейских популяциях, в котором было изучено 150 656 SNP. Полученные данные указывают на генетическую связь между миомой и злокачественными новообразованиями, а также на важность эстрогенов в развитии опухоли. Авторы провели метаанализ двух полногеномных исследований ассоциаций миомы у европейских женщин (16 595 гистологически подтвержденных случаев миомы и 52 330 контрольных). Согласно результатам метаанализа, в рост миом вовлечены как минимум два генетических пути, один из которых связан с онкогенезом (rs78378222 в TP53, rs10069690 в TERT, rs1800057 и rs1801516 в ATM, rs7907606 в OBFC1). В другом пути наиболее значимая ассоциация с миомой связана с низкочастотным вариантом 3'UTR в TP53, rs78378222 (p=4,03×10-37, регрессионный метаанализ, ОШ=1,74) [25].
Välimäki N. et al. провели полногеномное исследование ассоциаций у 15 453 больных c ММ, которые были идентифицированы на основании как самооценки фенотипа ММ, так и кодов Международной классификации болезней, и 392 628 контрольных пациентов в 2018 г. (изучено 805 426 SNP). Полногеномная статистика выявила rs117245733 в 13q14.11 как единственный SNP с предполагаемой (p<1010-5) ассоциацией как в когорте UKBB (ОШ=1,26; p=4,2×10-9), так и в когорте Хельсинки (ОШ=1,82; p=8,1×10-6). Гены, участвующие в стабильности генома, представлены TERT, TERC, OBFC1, TP53 и ATM. Гены, участвующие в развитии мочеполовой системы, – WNT4, WT1, SALL1, MED12, ESR1, GREB1, FOXO1, DMRT1 и маркерный антиген стволовых клеток матки CD44 образуют другую сильную подгруппу. Комбинированный риск по 22 локусам был связан с опухолями, положительными по мутации MED12 [26].
Edwards T.L. et al. в 2019 г. провели двухэтапный метаанализ генетических вариантов у женщин европейского и африканского происхождения на семи сайтах сети электронных медицинских карт и геномики (eMERGE) (3704 случая, подтвержденных методами визуализации, и 5591 контролей, подтвержденных методами визуализации), а также и женщин европейского и африканского происхождения из британского биобанка (UKBB, 5 772 случая и 61 457 контролей). Всего в метаанализ исследований ПГАА было включено 827 762 SNP. Окончательный анализ с 21 804 случаями и 205 525 контролями выявил 326 значимых для всего генома вариантов в 11 локусах, включая три новых локуса на хромосоме 1q24 (sentinel-SNP rs14361789; p=4,7×10-8), хромосоме 16q12.1 (sentinel-SNP rs4785384; p=1,5×10-9) и хромосоме 20q13.1 (sentinel-SNP rs6094982; p=2,6×10-8). Статистически значимые результаты дополнительно подтверждают ранее идентифицированные локусы, такие как SNP рядом с WT1, TNRC6B, SYNE1, BET1L и CDC42/WNT4. В нем сообщается о специфических по происхождению находках для сигнального SNP rs10917151 в локусе CDC42/WNT4 (p=1,76×10-24) [27].
Gallagher C.S. et al. в 2019 г. провели метаанализ ПГАА 35 474 случаев (подтвержденных либо на основе самоотчетов, либо клинически задокументированных случаев ММ) и 267 505 контрольных женщин европейского происхождения. В этом ПГАА было изучено 302 979 SNP; идентифицировано 8 новых значимых для всего генома p<5×10-8) локусов, ассоциированных с ММ (2p23.2, 4q22.3, 6p21.31, 7q31.2, 10p11.22, 11p14.1, 12q15 и 12q24.31), которые включают следующего кандидата интересующих генов: HMGA1, BABAM2 и WNT2. Помимо проверки 21 локуса, о котором сообщалось ранее, для 10 из них были обнаружены независимые сигналы. Фенотипическая стратификация ММ по тяжелым менструальным кровотечениям в 3409 случаях и 199 171 контрольной женщине показала полногеномные значимые отношения в трех из 29 локусов ММ: 5p15.33 (TERT), 5q35.2 (FGFR4) и 11q22.3 (ATM). Четыре локуса, обнаруженные в метаанализе, также связаны с риском эндометриоза; эпидемиологический метаанализ 402 868 женщин показывает, что у женщин с эндометриозом в анамнезе как минимум в два раза выше вероятность диагноза ММ [28].
Masuda T. et al. в 2020 г. использовали данные проекта Biobank Japan Project для проведения полногеномных ассоциативных исследований пяти гинекологических заболеваний (клинически диагностированных 5236 случаев ММ, 645 эндометриоза, 647 рака яичников, 909 рака эндометрия матки и 538 рака шейки матки, и 39 556 общих женских заболеваний); контроль включал 175 574 SNP. Анализы, проведенные в рамках логистической модели, линейной смешанной модели и модели, включающей корреляции, выявили четыре локуса для ММ (rs7412010, chr:g.22436446 G>C на 1p36, CDC42/WNT4, p=1,2×10-12; rs12415148, chr10:g.105680586 T>C на 10q24, STN1 (OBFC1), p=3,5×10-10; rs12225799, chr11:g.241124 C>G на 11p15, BET1L/RIC8A, p=1,1×10-21; rs17332320, chr22:g.40711620 G>T на 22q31, TNRC6B, p=1,6×10-12) [29].
Sakai K. et al. провела двухэтапное ПГАА в 2020 г. с 13 746 случаями ММ, диагностированными с помощью ультразвукового исследования и/или магнитно-резонансной томографии, и 70 316 контрольных пациентов без ММ и злокачественных новообразований в анамнезе из японской популяции с последующим повторным исследованием с 3483 случаями и 4795 контролями. В данном ПГАА изучено 951 117 SNP, обнаружено 9 значимых локусов, в том числе новый на 12q23.2 (rs17033114, p=6,12×10-25 с ОШ=1,177 (1,141–1,213), LINC00485). Анализ подгрупп показал, что 5 локусов (3q26.2, 5p15.33, 10q24.33, 11p15.5, 13q14.11) были статистически значимыми среди множественных миом и 2 локуса (3q26.2, 10q24.33) были значимы среди подслизистых миом. Плейотропный анализ показал, что все 9 локусов были связаны по крайней мере с одним пролиферативным заболеванием, что указывает на то, что эти гены играют роль в одном и том же неопластическом пути. Кроме того, аллель T риска rs2251795 (3q26.2) был связан с большей длиной теломер как в нормальных, так и в опухолевых тканях [30]. Кроме того, имеются противоречивые данные о том, что как в кариотипически нормальных, так и в аномальных образцах миом теломерные повторы в среднем на ~40% короче, чем в соответствующем миометрии [31]. Полученные данные проливают свет на важную роль генетических причин в этиологии миомы.
За период с 2011 г. по настоящее время (2022 г.) выполнено 8 полногеномных исследований ММ (табл. 1), в результате которых выявлено 34 гена и 79 ПГАА-значимых однонуклеотидных полиморфизмов (SNP), ассоциированных с развитием ММ [23–30]. В таблице 2 представлены данные о вовлеченных в ММ генах, показана ассоциация с заболеванием в двух и более ПГАА (выявлено 19 генов и 62 ПГАА-значимых полиморфизма локусов, ассоциированных с риском развития ММ).
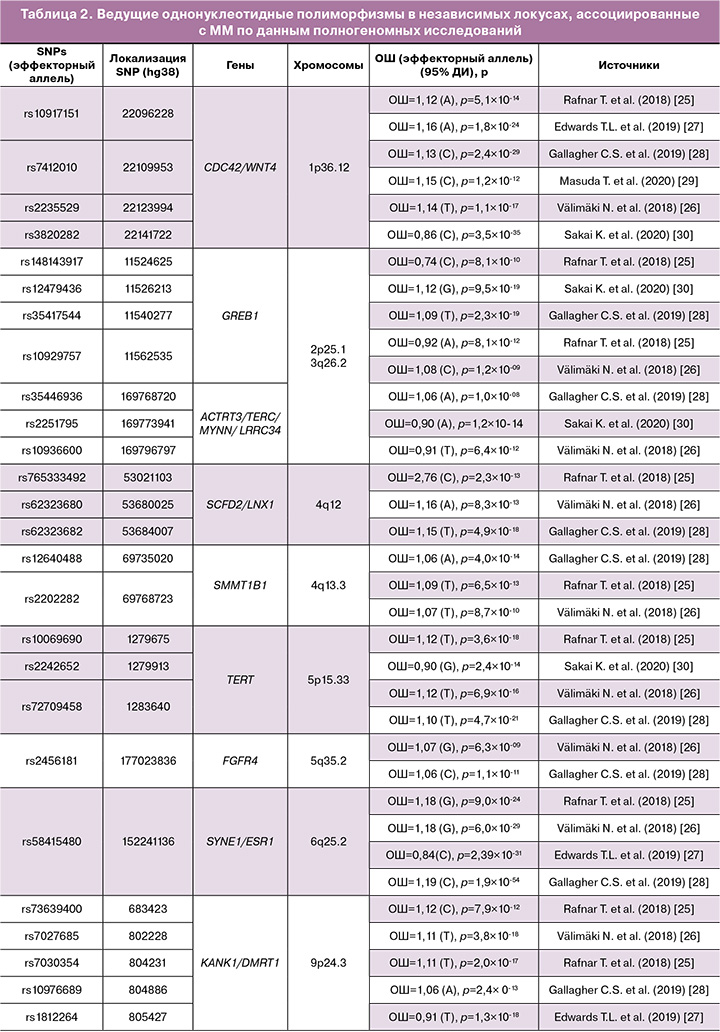
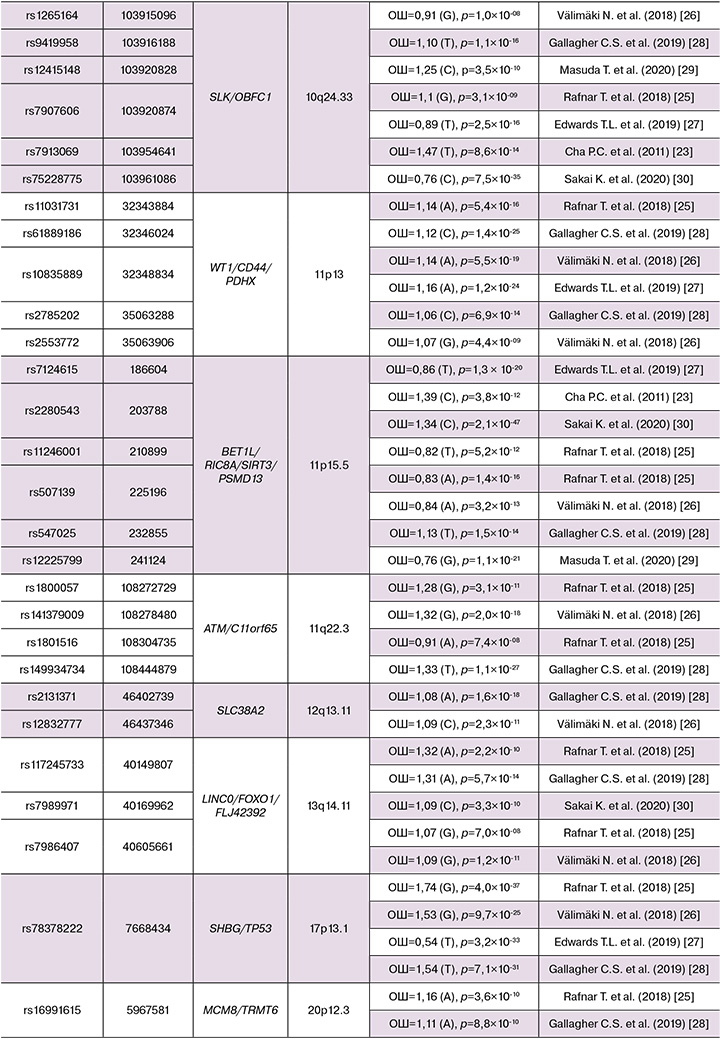
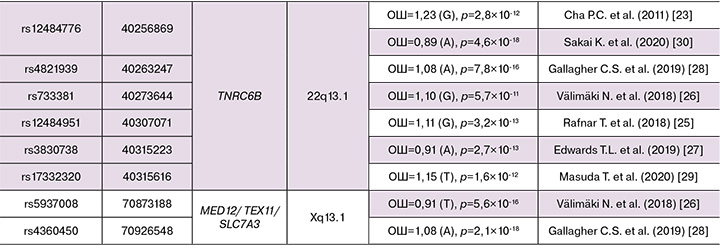
Полногеномные исследования ММ проводились среди жителей Японии (2 ПГАА), Европы (3 ПГАА) и др. В опубликованных полногеномных исследованиях ММ и некоторых распространенных однонуклеотидных полиморфизмов (SNP), связанных с риском миомы, некоторые гены и их близлежащие сайты были реплицированы: в 7 исследованиях расположены в 10q24.3 (SLK/OBFC1) [23, 25–30], 11p15.5 (BET1L/RIC8A/SIRT3/PSMD13) [23, 25–30], 22q13.1 (TNRC6B) [23, 25–30], в 6 исследованиях: 1p36.12(CDC42/WNT4) [25–30], в 4 исследованиях: 2p25.1 (GREB1) [25, 26, 28, 30], 5p15.33 (TERT) [25, 26, 28, 30], 6q25.2 (SYNE1/ESR1) [25–28], 9p24.3 (KANK1/DMRT1) [25–28], 11p13 (WT1/CD44/PDHX) [25–28], 13q14.11 (LINC0/FOXO1 /FLJ42392) [25, 26, 28, 30], 17p13.1 (SHBG/TP53) [25–28], в 3 исследованиях: 3q26.2 (ACTRT3/TERC/MYNN/LRRC34) [26, 28, 30], 4q12 (SCFD2/LNX1) [25, 26, 28], 4q13.3 (SММT1B1) [25, 26, 28], 11q22.3 (ATM/C11orf65) [25, 26, 28], в 2 исследованиях: 5q35.2 (FGFR4) [26,28], 12q13.11 (SLC38A2) [26, 28], 20p12.3 (MCM8/TRMT6) [25,28], Xq13.1 (MED12/TEX11/SLC7A3) [26, 28].
Следует отметить, что 15 из 34 известных на сегодняшний день ПГАА-значимых генов показали ассоциацию с заболеванием только в одном полногеномном исследовании, что, безусловно, недостаточно и диктует необходимость дальнейших репликативных исследований для установления роли этих генов в возникновении ММ. Кроме того, ММ содержит генетические изменения некоторых генов-драйверов, включая мутации MED12, биаллельную инактивацию FH и перестройки HMGA2 [32, 33]. Однако в настоящее время только часть генетических вариаций ММ может быть объяснена.
Патогеномика ММ предлагает новые взгляды на профилактику, диагностику и лечение этого заболевания. Для лечения ММ со сверхэкспрессией гена HMGA2 особенно перспективным является ингибирование сигнального пути, связанного с активацией гена инсулиноподобного фактора роста 2 мРНК-связывающего белка 2 (IGF2BP2). Новым и перспективным подходом к фармакотерапии ММ является эффективное управление его размером (супрессия генов TGF- и ACVR1). В настоящее время появляется больше возможностей для определения вероятности развития ММ и особенностей его клинического проявления благодаря обнаружению генов-кандидатов заболевания, а также нежелательных сочетаний минорных аллелей и их эпигенетической регуляции. Таким образом, можно разработать инновационные подходы к лечению этого важного гинекологического заболевания, а также стратегии его ранней профилактики [34].
Однонуклеотидные полиморфизмы (SNP) также были реплицированы в 4 исследованиях, включая rs58415480 [25–28] и rs78378222 [25–28]. Большинство из них были повторены в 2 исследованиях, таких как rs10917151 [25, 27], rs7412010 [28, 29], rs10929757 [25, 26], rs2202282 [25, 26], rs72709458 [26, 28], rs2456181 [26, 28], rs7907606 [25, 27], rs10835889 [26, 28], rs2280543 [23, 30], rs507139 [25, 26], rs117245733 [25, 28], rs7986407 [25, 26], rs16991615 [25, 28] и rs12484776 [23, 30]. Таким образом, в полногеномных исследованиях были реплицированы 16 SNP из 79 ПГАА-значимых локусов, что может свидетельствовать о достоверности данных об ассоциации этих полиморфизмов с ММ. Кроме того, эти 16 полиморфных ПГАА-значимых SNP, реплицированных для ММ, могут быть рекомендованы для дальнейших репликативных исследований в других популяциях. Вероятность подтверждения их связи с заболеванием у различных этно-территориальных групп населения достаточно высока. Важно отметить, что подавляющее большинство ассоциированных с ММ локусов (63 из 79 известных на сегодняшний день) показали ассоциацию с заболеванием только в одном ПГАА, и, конечно, для этих локусов необходимы дополнительные репликативные исследования, которые подтвердят или опровергнут их связь с болезнью.
Во многих исследованиях, наряду с применением ПГАА, было выполнено дополнительное секвенирование экзонов. В одном исследовании, проведенном Яценко С.А. и соавт., для изучения геномного ландшафта ММ использовались полноэкзомное секвенирование и геномные массивы. Это исследование включало анализ 16 недавно замороженных образцов миомы и соответствующих неповрежденных тканей миометрия, а также 153 образцов миосарком матки, полученных от женщин, у которых была диагностирована миома или миосаркома матки и которым в рамках клинического лечения была проведена абдоминальная гистерэктомия. Авторы идентифицировали несколько повторяющихся генетических изменений в генах, участвующих в ремоделировании хроматина, клеточной пролиферации и клеточных сигнальных путях. Они также обнаружили, что геномный ландшафт ММ очень гетерогенен, MED12-отрицательные миомы включают изменения числа копий, влияющие на компоненты медиаторного комплекса, такие как MED8, MED18, CDK8 и длинную межгенную небелковую кодирующую РНК 340 (CASC15), которые могут влиять на архитектуру медиатора и/или транскрипционную активность. Авторы обнаружили мутации, связанные с миомагенезом, в генах COL4A6, DCN и AHR, а также в новых генах NRG1, ADAM18, HUWE1, FBXW4, FBXL13 и CAPRIN1 [35].
Kämpjärvi K. et al. в дополнение к ПГАА использовали целевое секвенирование экзонов для изучения генетических мутаций в ММ у большой когорты женщин. 611 опухолей, отрицательных по мутации экзона 2 MED12, были проанализированы на наличие потенциальных мутаций экзона 1 MED12. Авторы обнаружили, что большинство ММ имели мутации в гене субъединицы 12 медиаторного комплекса (MED12), который участвует в регуляции транскрипции, поскольку наблюдались пять мутаций, которые были вставками/делециями в рамке считывания. Согласно данным экспрессии в масштабе всего транскриптома, мутации экзона 1 и экзона 2 MED12 приводят к одному и тому же отчетливому глобальному паттерну экспрессии генов, при этом RAD51B является наиболее повышенным геном. Мутации экзона 1 и экзона 2 нарушают связь между MED12, циклином C и CDK8/19 и удаляют связанную с медиатором активность киназы CDK, согласно экспериментам по иммунопреципитации и активности киназы. Полученные данные подчеркивают важность MED12 при ММ и демонстрируют, что экзон 1 и экзон 2 имеют схожие онкогенные эффекты, а также подчеркивают необходимость включения экзона 1 в будущие исследования MED12 [36].
В другом исследовании, проведенном Firdaus R. et al., целевое секвенирование экзонов использовалось для изучения генетических мутаций в ММ у группы немецких женщин. В данном исследовании 22 множественные фибромиомы из одной и той же матки и из разных маток 4 женщин были протестированы на наличие соматических мутаций в регионах «горячих точек» MED12. Авторы обнаружили, что большинство ММ имеют мутации в гене субъединицы 12 медиаторного комплекса (MED12), который участвует в регуляции транскрипции. В частности, они выявили множественные мутации в экзоне 2 гена MED12, которые были связаны с более высокой скоростью роста лейомиомы, и фланкирующие интронные области, семь экзонных вариантов и пять интронных вариантов, которые предоставляют доказательства того, что множественные ММ в одной матке могут быть не клонального происхождения. В исследовании также сравнивались профили мутаций ММ у немецких женщин с профилями из других популяций, и было обнаружено, что характер мутаций в разных этнических группах был сходным [37].
В этом исследовании, проведенном Ajabnoor G.M. et al., использовали направленное секвенирование экзонов для исследования генетических мутаций в ММ у когорты женщин из Саудовской Аравии. Всего в отделении гистопатологии KAUH, Джидда, было собрано 154 образца ткани миомы и миометрия, соответствующих 77 образцам матки, полученным при гистерэктомии. Авторы обнаружили, что большинство ММ имеют мутации в гене субъединицы 12 медиаторного комплекса (MED12), который участвует в регуляции транскрипции. Они отметили, что более 44% (34/77) миом арабских женщин имеют различные мутации MED12 (30 миссенс-мутаций, 1 участок сплайсинга и 3 вставки и удаления). Они обнаружили новые соматические мутации в кодонах 36, 38 и 55 в дополнение к известному кодону 44. Большинство генетически модифицированных опухолей (27/30; 90%) показали только один вид генетических изменений, демонстрируя, что даже один сдвиг аллеля в MED12 может иметь существенное влияние на трансформацию нормального миометрия в миомы. Когда опухоль положительна на мутацию MED12 (p<0,05), наблюдается интригующая обратная связь между размером опухоли и ЛГ. Компьютерные исследования показывают, что мутации аминокислотных замен в области экзона-2 MED12 могут привести к возможным изменениям фенотипа и стабильности белка. В исследовании также сравнивались профили мутаций ММ у женщин из Саудовской Аравии и у женщин из других популяций. Было обнаружено, что модели мутаций были схожими в разных этнических группах. Однако авторы отметили, что частота и распределение мутаций MED12 различались между разными исследованиями и популяциями [38].
В исследовании, проведенном Bertsch E. et al., использовалось целевое секвенирование экзонов для изучения генетических мутаций в ММ и миосаркомах. Всего для исследования было отобрано 178 пациенток с гистологическим диагнозом «обычная миома» и 32 пациентки с миосаркомой матки. Было обнаружено, что мутации MED12 присутствовали в 74,7% (133/178) миом, что согласуется со многими независимыми исследованиями. Для сравнения, мутации MED12 были обнаружены всего в 9,7% (3/32) миосарком. Эти данные показывают, что MED12 может играть функциональную роль, как в доброкачественных, так и в злокачественных опухолях гладкой мускулатуры матки. Конкретные обнаруженные мутации различались для каждой опухоли, однако большинство мутаций MED12 были обнаружены в экзоне 2 гена MED12. При дальнейшем изучении экспрессии HMGA2 во всех миомах и миосаркомах было показано, что сверхэкспрессия HMGA2 присутствует исключительно в миомах без мутации MED12, составляя 10,1% (18/178) от общего числа миом и 40% (18/45) от миом без мутации MED12. Сверхэкспрессия HMGA2 наблюдалась в 25% (8/32) миосарком, а в HMGA2-положительных миосаркомах не было обнаружено мутаций MED12. Эти данные ясно указывают на то, что мутации MED12 и сверхэкспрессия HMGA2 являются независимыми генетическими событиями в миомах и что они могут играть отдельные роли в формировании ММ [39].
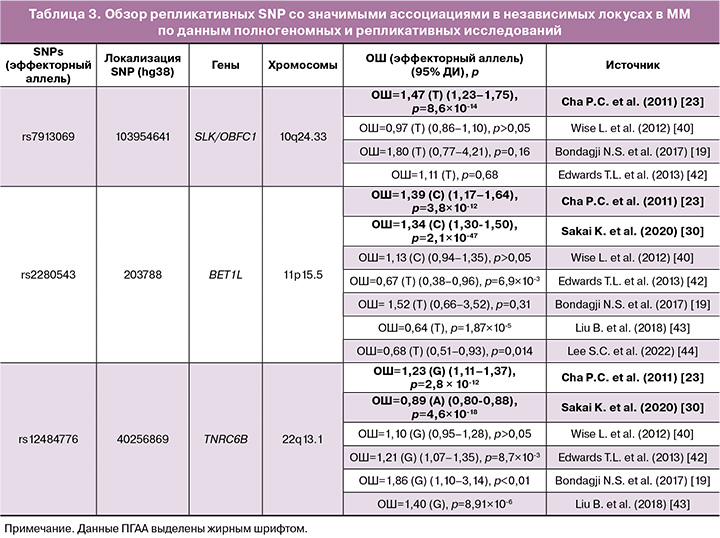
Также проводятся репликативные ПГАА-исследования значимых локусов, ассоциированных с развитием ММ. В частности, исследование Wise L. et al. (2012) [40] было посвящено изучению генетических факторов ММ у афроамериканок (проведено генотипирование более 1500 этнически значимых генетических маркеров). В выборке из 2453 пациенток с ММ, подтвержденными ультразвуковым исследованием или хирургическим путем, и 2102 участниц контрольной группы (Black Women's Health Study (BWHS)) было установлено участие в формировании ММ трех полиморфных локусов отдельных участков трех хромосом 2q37 (rs7573626), 4p16.1 (rs9715724) и 10q26 (rs7100028). Следует отметить, что ассоциации трех полиморфных локусов с развитием заболевания, ранее показанные в ПГАА-исследовании Cha P.C. et al. (2011) для японской популяции [23], не были подтверждены в данном исследовании [40].
Репликативное исследование трех ПГАА-значимых полиморфных локусов ММ, установленных ранее для японской популяции Cha P.C. et al. (2011) [23], было проведено Edwards T.L. et al. (2013) среди европейцев Америки [41]. В выборке американских женщин европеоидной расы, включающей 1086 пациенток с ММ, диагностированными с помощью визуализации малого таза, и 1549 человек в контрольной группе, сформированной из двух когорт (Right from the Start (RFTS) и биобанка ДНК BioVU), выявлена ассоциация развития ММ с двумя SNP – rs12484776 гена TNRC6B и rs2280543 гена BET1L. Ассоциация rs12484776 гена TNRC6B с объемом миоматозных узлов и rs2280543 гена BET1L с развитием интрамуральных миоматозных узлов была показана Edwards T.L. et al. (2013) [42].
В исследовании, проведенном Bondagji N.S. et al. в 2017 г. среди саудовских женщин, выполнено генотипирование 105 пациенток с ММ, диагностированных с помощью бимануального исследования, ультразвуковой визуализации и гистопатологического подтверждения биоптатов матки, полученных при гистерэктомии, и 112 здоровых контрольных женщин по пяти генетическим полиморфизмам; также исследовали силу корреляции между частотами генотипа и аллеля с риском развития ММ. Результаты показали, что у саудовских женщин с ММ была высокая распространенность аллеля G rs12484776 по сравнению с контрольной группой (ОШ 1,86; 95% ДИ 1,10–3,14; р<0,01). Авторы работы показали, что лица с генотипом AG по полиморфизму rs12484776 имеют в 2,6 раза больший риск развития ММ по сравнению с лицами с другими генотипами (ОШ 2,69; 95% ДИ 1,45–5,00; p<0,001). Не было показано, что частоты распределения генотипов для rs7913069 и rs2280543 повышают риск заболевания (для всех тестов p>0,05) [19].
Исследование, проведенное в 2018 г. Liu B. et al., было направлено на воспроизведение двух первоначальных важных генетических факторов, TNRC6B и BET1L, в популяции ханьцев. В общей сложности было набрано 674 женщины с ММ, диагностированной с помощью ультразвукового исследования, и 1381 человек, из которых были отобраны и генотипированы 55 SNP, картированных в TNRC6B и BET1L. Ассоциации между целевыми SNP и соответствующими клиническими признаками ММ были проанализированы только среди случаев. Два SNP, rs2280543 от BET1L (χ2=18,3; ОШ=0,64; p=1,87×10-5) и rs12484776 от TNRC6B (χ2=19,7; ОШ=1,40; p=8,91×10-6), были идентифицированы как значимо связанные с ММ. Rs2280543 значимо ассоциировался с количеством узлов миомы (p=0,0007), тогда как rs12484776 – с размером узлов (χ2=54,88; p=3,44×10-11). Оба SNP были значительными eQTL для своих генов [43].
В исследовании Lee S.C. et al. (2022) была впервые воспроизведена связь между BET1L rs2280543 и ММ у тайваньских женщин. Всего для исследования были отобраны 341 пациентка с ММ и 1656 лиц контрольной группы. Авторы интегрировали оцифрованные данные из базы данных Тайваньского биобанка (TWB) в медицинские записи участников (собраны диагностированные заболевания, генотип, образ жизни и биохимические данные) из Национальной базы данных исследований медицинского страхования (NHIRD). Участники с генотипом BET1L rs2280543 CT/TT имели ОШ=0,69 с 95% ДИ 0,51–0,93 по сравнению с участниками с генотипом BET1L rs2280543 CC (дикий тип). Вегетарианская диета и ММ не имели достоверной связи: ОШ=1,09 с 95% ДИ 0,77–1,55. С другой стороны, тест взаимодействия между rs2280543 и вегетарианской диетой был значимым (p=0,046). По сравнению с лицами с генотипом СС у вегетарианцев с генотипом СТ/ТТ была снижена частота ММ: ОШ=0,15 с 95% ДИ 0,05–0,47. Авторы обнаружили, что генотип BET1L rs2280543 CT/TT был связан со снижением заболеваемости ММ, особенно среди вегетарианцев [44].
Следует отметить, что различия в полученных результатах (ассоциации, подтвержденные в одних исследованиях, но не подтвержденные в других) могут быть вызваны уникальной генетической структурой изучаемых популяций, определяемой их этническими характеристиками, уникальными особенностями того, как факторы риска окружающей среды (питание и т.д.) влияют на этих людей, и другими факторами. Кроме того, необходимо продолжить репликативные исследования ММ с целью выявления локусов, определяющих предрасположенность женщин определенной этно-территориальной группы (популяции) к развитию этого заболевания.
Результаты указанных выше репликативных исследований обобщены в таблице 3. Проведенные ПГАА и репликативные исследования указывают на важнейшую роль в развитии ММ полиморфизмов rs2280543 гена BET1L (11p15.5), rs12484776 TNRC6B (22q13.1) (табл. 3). Полиморфный локус rs2280543 гена BET1L (11p15.5) был ПГАА-значимым в двух исследованиях [23, 30], и было подтверждено, что он связан с заболеванием в трех повторных исследованиях [42–44]. При этом аллель С rs2280543 оказался фактором риска развития ММ, а аллель Т этого полиморфизма – защитным фактором (табл. 3). В двух репликативных исследованиях ассоциация этого полиморфизма с ММ не была подтверждена [19, 40]. Полиморфизм rs12484776 TNRC6B (22q13.1) также был связан с ММ в двух полногеномных исследованиях [23,30] и воспроизведен в трех независимых исследованиях [19, 42, 43]. Вариант аллеля G rs12484776 TNRC6B (22q13.1) был ассоциирован с повышенным риском развития ММ, а аллель А оказался защитным фактором в отношении этого заболевания (табл. 3). В одном из исследований не было обнаружено статистически значимых ассоциаций этого полиморфизма с ММ [40]. Локус rs7913069 SLK/OBFC1 (10q24.33) хотя и является ПГАА-значимым для ММ [23], но ни одно из трех проведенных репликативных исследований не подтвердило его ассоциации с заболеванием [19, 40, 42].
Заключение
В обзоре рассмотрены основные полногеномные исследования ММ и определены ПГАА-значимые полиморфизмы, ассоциированные с ММ. Полученные данные о ПГАА-значимых локусах могут быть использованы для отбора полиморфизмов в репликативных исследованиях ММ в различных популяциях, а также для углубления понимания молекулярно-генетических механизмов развития заболевания. В результате секвенирования экзонов показано участие ряда генов, таких как MED12, в формировании ММ, что существенно расширяет представления о генетических детерминантах заболевания.


