
Введение
Инсульт – вторая по частоте причина смерти во всем мире. В мире ежегодно у 75 млн человек развивается инсульт, при этом 5 млн погибают, более 35 млн живут с последствиями инсульта. Ишемический инсульт (ИИ) – наиболее распространенный его тип, на долю которого приходится почти 80% всех зарегистрированных случаев острых нарушений мозгового кровообращения.
Научные достижения последних лет внесли большой вклад в понимание патобиологии и биохимических путей повреждения центральной нервной системы (ЦНС) при развитии ИИ. Клеточная смерть в нервной системе при нормальных физиологических условиях считается охранительной моделью. В процессе развития мозга нейроны в ЦНС подвергаются удалению, что помогает формировать нейронные связи и развивающуюся нейронную архитектуру мозга, т.е. запрограммированная гибель клеток играет важную роль в процессе развития нервной системы. Напротив, при достижении возраста зрелости и в течение всей последующей жизни нейроны подвергаются множеству травмирующих воздействий, что определяет их судьбу и выживание. Смерть клеток при острой локальной ишемии мозга включает начальную волну острого некротизирующего повреждения ткани мозга с последующим вторичным биохимическим повреждением, вызванным сложными биохимическими механизмами окружающей клеточной среды, что приводит к более организованной или запрограммированной форме гибели клеток. В последние годы в литературе описывалось несколько форм гибели клеток, которые происходят в клетке, сопровождаемой различными фенотипическими и молекулярными маркерами в зависимости от характера повреждения, испытываемого клеткой [1, 2].
Характеристика типов клеточной смерти
Клеточная смерть классифицируется на три типа: апоптотический (тип I), аутофагический (тип II) и некротический/онкотический (тип III). В зависимости от природы поражения смерть нейронов происходит по одному из этих механизмов, благодаря чему процесс может прогрессировать или прекращаться. Некроз и апоптоз нейронов были хорошо изучены. В последние годы исследователи получают все большее количество свидетельств о вовлеченности в процесс гибели клеток аутофагии и аутофагии-ассоциированной клеточной смерти. При этом для каждой формы клеточной смерти характерны свои биохимические маркеры (табл. 1, 2).
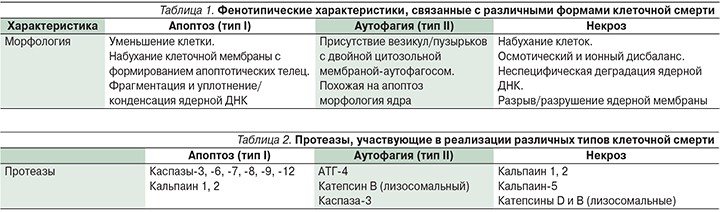
Некроз. Некроз традиционно рассматривается как неконтролируемая случайная форма клеточной смерти с морфологическими свойствами, отличными от апоптоза или аутофагии. Некротизируемая клетка в стандартном случае подвергается биоэнергетическому повреждению вследствие истощения АТФ, причиной которой служит воздействие повышения внутриклеточного Са2+, эксайтотоксичности или воспаления, сопровождается отеком внутриклеточных компонентов и нарушением целостности плазматической мембраны. Клеточный и ядерный лизис в результате некроза приводит к воспалению, что служит причиной дальнейшего повреждения области, окружающей зону некроза. Так, к некоторым из участвующих в биохимическом процессе некроза протеазы относятся кальций-зависимые кальпаины и лизосомальные катепсины, которые влияют на большинство внутриклеточных процессов.
В исследованиях последних лет получены данные, свидетельствующие о том, что некротический путь в действительности может быть регулируемым, зависящим от сигнализации некоторых вторичных внутриклеточных мессенджеров, однако эти результаты требуют дальнейшего подтверждения и описания точных биохимических процессов, их вызывающих. Одна из форм «запрограммированного» некроза, также называемая некроптозом, демонстрирует зависимость от взаимодействия с рецептором протеинкиназы RIP1, подавляемого ингибиторами RIP1, такими как некростатин [3].
Другим примером запрограммированного некроза служит активация поли-АДФ-рибозной полимеразы (PARP-1) с разрывом ДНК-цепей, что и считается причиной гибели клеток и наблюдается при определенных формах повреждения мозга.
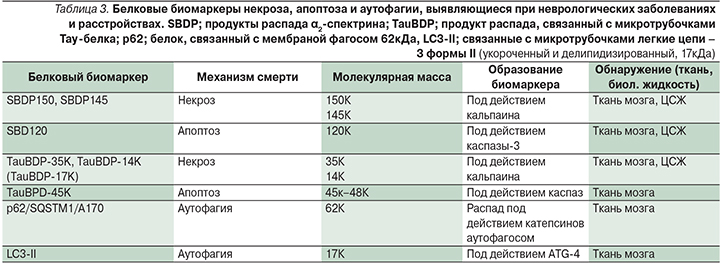
K.K. Wang обнаружил, что белок α2-спектрин, расположенный в аксонах, является отличным биомаркером некроза нейронов, т.к. последовательно расщепляется ферментом кальпаином на 2 разных фрагмента – SBDP 150 и BDP145 [4].
Апоптоз. Апоптоз – одна из хорошо изученных форм запрограммированной гибели клеток. Апоптоз играет меньшую роль в клеточной гибели при острых состояниях и наблюдается в более отдаленный период острой ишемии мозга (Е.И. Гусев). В апоптотической клетке описано развитие таких морфологических особенностей, как конденсация хроматина, ретракция псевдоподий, фрагментация ядра, набухание мембраны без выхода внутриклеточного содержимого во внеклеточное пространство. В зависимости от характера биохимической сигнализации, которой подвергается клетка, апоптоз можно классифицировать как: а) каспаз-зависимый и каспаз-независимый внутренний; б) внешний. Хотя дифференцировка этих путей основана на связанных с ними биохимических компонентах, существует множество перекрестных связей между путями посредством взаимодействия различных вовлеченных в этот процесс белков.
Внутренний апоптоз может быть запущен несколькими способами передачи сигналов, включая повреждение митохондрий, поражение эндоплазматического ретикулума, повреждение ДНК, окислительный стресс и избыток цитоплазмотического Ca2+. Особую роль в этом процессе отводят митохондриям. Потеря потенциала митохондриальной мембраной приводит к формированию повышения ее проницаемости и высвобождению митохондриальных белков – цитохрома С, вторичного активатора каспаз, полученного из митохондрий (SMAC), и высокотемпературного белка A2 (HTRA2) в цитозоль, а также к активации определенных проапоптотических митохондриальных мембранных белков, таких как Bid, которые инициируют развитие биохимических процессов, приводящих к активации проапоптотических каспазных протеаз, а также подавление антиапоптотических белков, выражающееся в формировании биохимических процессов, ведущих к индукции апоптоза. Не зависимый от каспазы апоптоз включает сигнализацию через белки, высвобождаемые из митохондриального межмембранного пространства (MIS), таких как апоптоз-индуцирующий фактор (AIF) и эндонуклеаза G, которые перемещаются в ядро, что приводит к необратимому повреждению ДНК.
Внешний апоптоз определяет форму гибели клеток, распространяемую через специфические трансмембранные рецепторы в ответ на внеклеточные стресс-сигналы. Сигналинг инициируется связыванием лиганда (Fas или TNF) с рецепторами смерти, которые вызывают конформационные изменения в цитоплазматическом домене рецепторов, и инициирует образование смерть-индуцирующего сигнального комплекса (DISC), который активирует каспазу-8, в свою очередь активирующую эффекторные каспазы, такие как каспаза-3, -6 и -7, что в конечном итоге приводит к апоптозу.
Стресс эндоплазматического ретикулума и развернутый белковый ответ апоптоза
Клеточная смерть, опосредуемая стрессом эндоплазматического ретикулума (ЭР) и включением реакции денатурации белков (UPR), предложена в качестве третьей формы апоптоза. Эти механизмы служат фактором, способствующим развитию хронических нейродегенеративных нарушений [5, 6].
Исследования выявили связь между ионами ЭР и митохондрий при гибели клеток. Белки Bcl-2, которые присутствуют на мембране эндоплазматического ретикулума, снова имеют критическое значение в последовательности событий, которые разыгрываются при активации пути ЭР-стресса. Известно, что стресс ЭР активируется каспазой-12, которая в дальнейшем может активировать каскад протеолитических эффекторных каспаз, приводя к гибели клеток [7].
Кроме того, другой продукт распада, α2-спектрина (SBDP120), образующийся под воздействием каспазы-3, в настоящее время используется в качестве биомаркера апоптоза нейронов как в тканях, так и биологических жидкостях, например в цереброспинальной жидкости – ЦСЖ (табл. 3) [8].
Аутофагия. Аутофагия при остром мозговом повреждении была продемонстрирована в эксперименте на грызунах. В современной литературе мнения насчет предполагаемой роли аутофагии в механизмах выживания и гибели клеток неоднозначны. Исследования выявили, что аутофагия нейронов – это тонко регулируемый процесс. Подавление процесса аутофагии и супрессия гена аутофагии (atg7) в ЦНС привели к обширной нейродегенерации [9]. Таким образом, аутофагия играет критическую роль в выживании постмитотических нейронов ЦНС.
Аутофагия была описана как внутриклеточный механизм, который активируется, когда клетки подвергаются внешнему воздействию, как, например, недостаточность питательных веществ или другие клеточные повреждения, такие как эксайтоксичность [10]. Аутофагию характеризует наличие везикул с двойной мембраной, называемых аутофагосомами, которые, как считается, происходят из ЭР, в пределах клетки изолирующих поврежденные органеллы, такие как митохондрии, в конце концов попадающие в лизосомы путем слияния наружной мембраны аутофагосом с лизосомальной мембраной, что приводит к образованию однослойных мембранных структур, называемых аутолизосомами, внутри которых лизосомальные гидролазы разрушают органеллы для рециркуляции простых аминокислот обратно в клеточный метаболизм для поддержания жизнеобеспечения клеток и гомеостаза.
Активация аутофагии в головном мозге отмечается при окклюзии сонной артерии и гипоксемии в экспериментах на грызунах [11, 12]. В исследованиях на неонатальных ишемических моделях экспериментального инсульта активация аутофагии имела отрицательное влияние на выживание нейронов [13]. В другом исследовании [14] был продемонстрирован нейропротективный эффект на нейроны гиппокампа вследствие истощения гена аутофагии atg7 на экспериментальной модели неонатальной гипоксии-ишемии. По всей видимости, данные результаты могут быть применимы только к новорожденным, поскольку дефицит в гене atg7 в более позднем возрасте приводит к дегенерации нейронов гиппокампа начиная с возраста около 3 недель. Экспериментальная ишемия у взрослых диких мышей (PND 56) вызывала активацию аутофагии, однако отрицательного эффекта на выживание нейронов отмечено не было. Тем не менее другие исследования с использованием индукторов аутофагии, таких как парамицин, выявили положительную роль аутофагии посредством активации сигнального пути Akt/CREB [15]. Результаты этих исследований предполагают, что ингибирование аутофагии может приводить к усилению гибели клеток [16].
Биомаркером аутофагии в ЦНС служит изоформа легкой цепи белка MAP-LC3 (или atg8). При нейрональной аутофагии или смерти клетки с помощью аутофагии появляется еще один маркер – белок p62. Этот белок индуцируется стрессом и является одним из белков, связывающих LC3, что приводит к образованию аутофагосом. Важно, что при активно происходящем процессе аутофагии уровень p62 снижается [17]. Таким образом, p62 является возможным маркером аутофагии при травмах и расстройствах в ЦНС.
Заключение
В настоящее время существует настоятельная потребность в создании диагностических тестов, основанных на простых биологических жидкостях, используемых для ведении пациентов с острой локальной ишемией, будь то мониторинг пациентов, находящихся в отделении интенсивной терапии, или прогнозирование степени тяжести повреждения ткани мозга в острейшей периоде ИИ уже на уровне приемного отделения стационара, а также могут быть полезными для назначения дифференцированной индивидуализированной терапии и реабилитационного лечения.
Биомаркеры будут обладать важными прогностическими функциями; способствовать разработке рекомендаций по скорейшему возвращению к службе или работе, а также предоставят возможность для консультирования пациентов, освобожденных от трудовой деятельности. Маркеры биохимических процессов открывают большие возможности для проведения клинических исследований, включая подтверждение эффективности целевой лекарственной терапии. Временный профиль изменений в биомаркерах будет определять сроки лечения. Биомаркеры служат большим подспорьем в клинических исследованиях, проводимых на ранних этапах заболеваний, и могут быть гораздо более надежными и экономичными средствами, чем обычные неврологические способы оценки. Таким образом, эти биомаркеры значительно уменьшат риски и затраты, связанные как с клиническими испытаниями, так и в конечном счете с лечением и ведением пациентов.


