
Введение
В настоящее время препаратом выбора для лечения анемии у пациентов с хронической почечной недостаточностью (ХПН) признаны эритропоэз-стимулирующие средства короткого и пролонгированного действий [1]. Молекула рекомбинантного человеческого эритропоэтина (рчЭПО) имеет высокое сродство к рецептору, что определяет выраженный биологический эффект in vitro [2, 3]. Однако in vivo рчЭПО быстро инактивируется печенью и элиминируется из организма, что определяет необходимость введения препарата до 3 раз в неделю [4]. Поскольку терапия аналогами эндогенного эритропоэтина проводится в течение длительного времени, а в ряде случаев пожизненно, частота выполнения инъекций определяет качество жизни пациентов и их приверженность лечению. Известно, что дополнительное гликозилирование (присоединение дополнительных углеводных цепей) молекулы эритропоэтина приводит к увеличению ее периода полувыведения и как следствие – к более продолжительному биологическому эффекту. Дарбэпоэтин альфа был создан путем присоединения двух дополнительных углеводных цепей к молекуле рчЭПО, при этом меньшее сродство новой молекулы к рецептору эритропоэтина компенсируется ее более высокой активностью и длительной циркуляцией в крови [2].
Другим способом увеличения периода полувыведения белковой молекулы служит присоединение к ней молекулы полиэтиленгликоля (ПЭГ). ПЭГ является индифферентным для организма веществом, присоединение его к рчЭПО способствует увеличению периода полувыведения, что позволяет уменьшить кратность инъекций препарата, а увеличение гидродинамического радиуса молекулы обусловливает снижение иммуногенности белка. При этом молекулярная масса ПЭГ определяет баланс фармакокинетических характеристик и активности молекулы, поскольку присоединение ПЭГ с небольшой молекулярной массой значимо не изменяет фармакокинетику молекулы, а присоединение ПЭГ со слишком большой молекулярной массой может приводить к снижению активности препарата. Оптимальной считается молекулярная масса ПЭГ около 30 кДа. В настоящее время единственным препаратом пегилированного эритропоэтина является C.E.R.A. (continuous erythropoietin receptor activator – активатор рецепторов эритропоэтина длительного действия), обладающий бόльшим по сравнению с дарбэпоэтином периодом полувыведения [5].
ЗАО «БИОКАД» разработан инновационный оригинальный препарат пегилированного дарбэпоэтина – BCD-131. Идеология создания новой молекулы включила оба описанных выше подхода: гликолизирование и пегилирование эритропоэтина [6].
В рамках доклинических исследований детально изучены структурные, физико-химические и биологические свойства нового биотехнологического продукта. Показано, что препарат BCD-131 обладает специфической активностью in vitro и in vivo: повышает уровень гемоглобина и увеличивает количество эритроцитов в крови путем избирательного воздействия на рецепторы эритропоэтина. Результаты исследований острой и хронической токсичности продемонстрировали отсутствие гибели экспериментальных животных, значимого изменения их общего состояния, функционирования органов и систем, местно раздражающего действия и иммуногенности (табл. 1).

Первым этапом клинической разработки препарата BCD-131 стала оценка фармакокинетики, фармакодинамики, переносимости, безопасности и иммуногенности при его однократном подкожном введении в возрастающих дозах здоровым добровольцам в рамках исследования I фазы BCD-131-1 (NCT02731469). На основании полученных результатов был определен диапазон терапевтических доз исследуемого препарата BCD-131: 1,05 мкг/кг, 1,7 мкг/кг и 2,75 мкг/кг, а также подтверждена возможность его введения один раз в месяц [7].
Целью исследования II фазы BCD-131-2 (NCT03519243) стало определение терапевтически эффективной и безопасной дозы препарата BCD-131 при его многократном применении в лечении анемии у больных ХПН, находящихся на диализе. Результаты данного исследования представлены в настоящей статье.
Материалы и методы
Дизайн исследования: международное многоцентровое рандомизированное открытое сравнительное клиническое исследование.
Критерии включения. В исследовании принимали участие мужчины и женщины в возрасте от 18 до 75 лет включительно с терминальной стадией ХПН, в течение как минимум последних 3 месяцев до подписания информированного согласия (ИС), получавшие диализ (не менее 12 часов в неделю, индекс диализной дозы [Kt/v]≥1,2 для пациентов, получавших гемодиализ, и Kt/v≥1,7 для пациентов, получавших перитонеальный диализ) и препараты рчЭПО. В течение 2 недель до подписания ИС уровень гемоглобина у пациентов должен был поддерживаться в пределах целевых значений (100–120 г/л) при введении стабильной дозы рчЭПО. На скрининге коэффициент насыщения трансферрина должен был составлять 20% и более, уровень ферритина – более 100 нг/мл. В исследование не включались пациенты с анемией другой, не почечной, этиологии, рядом острых и хронических заболеваний, в том числе волчаночным нефритом, системным васкулитом, а также при наличии определенных протоколом лабораторных отклонений на скрининге.
Набор пациентов осуществлен в Российской Федерации в 13 исследовательских центрах, в Республике Беларусь – в 3 исследовательских центрах.
До начала любых процедур пациентам была предоставлена полная информация, необходимая для принятия осознанного и взвешенного решения об участии в данном клиническом исследовании. После подписания информированного согласия (ИС) участнику проводилось скрининговое обследование для подтверждения соответствия критериям отбора в исследование.
Осуществлялась стратификация участников в соответствии с видом предшествовавшей терапии (эпоэтин альфа/эпоэтин бета/дарбэпоэтин альфа), а также в зависимости от наличия факторов риска развития нежелательных явлений (НЯ) на фоне терапии рчЭПО: возраст (<60/≥60 лет), наличие/отсутствие сосудистых протезов, необходимость/отсутствие необходимости применения гипогликемических препаратов или инсулина.
Первоначально было запланировано включение в исследование 4 групп пациентов: 3 группы исследуемого препарата BCD-131 (1,05 мкг/кг, 1,7 и 2,75 мкг/кг) и группа препарата сравнения Мирцера. Индивидуальная ежемесячная доза исследуемого препарата BCD-131 рассчитывалась исходя из дозы рчЭПО/дарбэпоэтина альфа, вводившейся пациенту до подписания ИС, с применением конверсионного коэффициента (КК) 0,8, выбранного на основании анализа обобщенных данных клинических исследований препарата Мирцера (табл. 2).
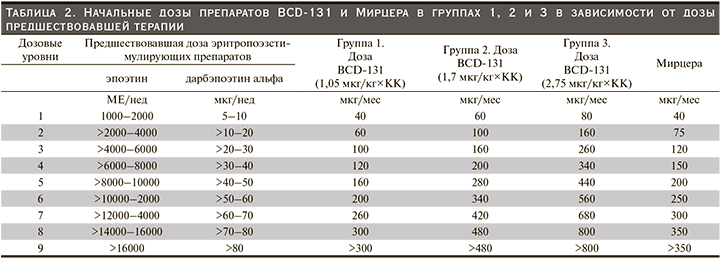
Исследование состояло из двух этапов. На первом этапе пациентов включали последовательно в группы исследуемого препарата и параллельно с этим производился набор участников в группу препарата сравнения в соотношении 3:1. Сначала в первую группу, BCD-131 1,05 мкг/кг, были включены 9 пациентов, в группу Мирцера – 3 участника. После того как все пациенты получили по две инъекции исследуемого препарата/препарата сравнения и прошли 2-недельный период наблюдения после второй инъекции, на основании полученных данных о фармакодинамике и безопасности препарата BCD-131 при его применении в дозе 1,05 мкг/кг специально созданная комиссия (Комитет по обеспечению безопасности дозирования когорт пациентов [КОБ] приняла решение о возможности эскалации дозы и включении следующих 9 пациентов во вторую группу, BCD-131 1,7 мкг/кг, и еще 3 участников в группу препарата Мирцера. После получения всеми вновь включенными пациентами 2 инъекций исследуемого препарата/препарата сравнения и завершения 2-недельного периода наблюдения КОБ рассмотрел полученные данные и установил, что при введении исследуемого препарата BCD-131 в дозах 1,05 мкг/кг и 1,7 мкг/кг наблюдается достаточный фармакодинамический и клинический эффекты, что определяет отсутствие необходимости дальнейшей эскалации дозы исследуемого препарата. Лечение всех включенных в исследование пациентов не прерывалось и продолжалось в прежней дозе. Исследуемый препарат и препарат сравнения вводили 1 раз в месяц подкожно.
На втором этапе в каждую из сформированных на первом этапе групп дополнительно были включены пациенты до общего числа участников в каждой группе 25 человек, включая рандомизированных на первом этапе. Пациенты продолжали терапию вплоть до 21-й недели. Длительность периода наблюдения составила 28 дней после последнего введения исследуемого препарата/препарата сравнения.
Первичная конечная точка для оценки эффективности: изменение уровня гемоглобина – разница среднего арифметического уровня гемоглобина на неделях 21 и 23 и исходного уровня гемоглобина (среднее арифметическое уровня гемоглобина на скрининговом визите и визите 1). Вторичные конечные точки: доля пациентов с целевым уровнем гемоглобина (100–120 г/л) и средний уровень гемоглобина на протяжении 23 недель, доля участников, которым потребовалось проведение коррекции дозы исследуемого препарата/препарата сравнения. Итоговый статистический анализ эффективности проведен в популяции per-protocol, включившей 65 пациентов, прошедших запланированный курс лечения и завершивших участие в исследовании в соответствии с протоколом.
Конечными точками для оценки безопасности стали доля пациентов, у которых зарегистрировано развитие нежелательных явлений/серьезных нежелательных явлений (НЯ/СНЯ), НЯ 3–4-й ст. тяжести, в том числе связанных с исследуемой терапией, а также доля пациентов в каждой группе, досрочно прекративших участие в исследовании в связи с развитием НЯ/СНЯ. Степень тяжести НЯ и лабораторных отклонений оценивалась в соответствии с классификацией СТСАЕ v. 4.03 (Common Terminology Criteria for Adverse Events) [8]. Анализ безопасности проведен в популяции пациентов, получивших по крайней мере одну дозу исследуемого препарата/препарата сравнения (n=75).
Конечной точкой для оценки иммуногенности служила доля пациентов с выявленными связывающими (САТ) и нейтрализующими (НАТ) антителами к пегилированному дарбэпоэтину/метоксиполиэтиленгликоль-эпоэтину бета. Сначала при помощи валидированных методов твердофазного иммуноферментного анализа (ELISA) проводилась оценка наличия САТ (скрининговый и подтверждающий анализы). В случае обнаружения САТ предполагалось выполнение клеточного теста для определения НАТ. В анализ иммуногенности были включены 74 участника, получившие хотя бы одно введение исследуемого препарата/препарата сравнения, у которых имелись пригодные для анализа образцы, взятые до первого введения препаратов и как минимум на одном из последующих визитов (на неделях 9 и 23).
Для расчета размера выборки были использованы результаты многоцентрового рандомизированного открытого клинического исследования препарата Мирцера в рамках поддерживающей терапии анемии у пациентов с ХПН, находившихся на диализе [9, 10]. В качестве критерия эффективности были использованы данные об изменении уровня гемоглобина у пациентов на фоне терапии. Расчет популяции исследования был произведен на основании метода, описанного Chow Sh.-Ch., Shao J., Wang H., 2008 [11]. Учитывая необходимость оценки первичной терапевтической эффективности и выбора дозы исследуемого препарата BCD-131 для последующего изучения, а также высокую вероятность досрочного выбывания пациентов из исследования (в связи с характером основного заболевания), для получения релевантных данных в каждую группу были включены по 25 пациентов.
Гипотеза об эквивалентной эффективности исследуемого препарата и препарата сравнения была проверена при следующих значениях ошибок: ошибка первого рода – 5% (α=0,05), ошибка второго рода – 20% (β=0,2), мощность критерия – 80%. Граница эквивалентности исследуемого препарата и препарата сравнения была принята равной 10 г/л как половина диапазона целевых значений гемоглобина 100–120 г/л.
Рандомизация
Рандомизация была центральной. На первом этапе пациенты включались в группу 1 исследуемого препарата BCD-131 1,05 мкг/кг и группу препарата сравнения Мирцера в соотношении 3:1. После принятия решения об эскалации дозы препарата BCD-131 по такому же принципу, в соотношении 3:1, были набраны пациенты во вторую группу исследуемого препарата BCD-131 1,7 мкг/кг и в группу препарата сравнения. Дальнейшей эскалации дозы не потребовалось. На втором этапе проводился дополнительный набор пациентов во все группы терапии до 25 человек в каждой. Исследователю сообщались только идентификационный номер пациента и группа терапии.
Статистический анализ производился с помощью программной среды SAS 9.4 и языка программирования для статистической обработки данных R. Выбор метода описательной статистики и статистического сравнения определялся типом данных и видом распределения. Для описания количественных переменных, распределенных по нормальному закону, использовались средние значения и стандартные отклонения, для статистической обработки – двухвыборочный критерий Стьюдента и дисперсионный анализ. Для количественных данных, распределенных по отличному от нормального закону распределения, средние значения описывались с помощью медиан и интерквартильных размахов; для статистической обработки применялись критерии Краскела–Уоллиса и Манна–Уитни с поправкой Беньямини–Йекутили. Для описания категориальных данных использовались проценты или доли. Статистическое сравнение категориальных данных проводилось с использованием точного теста Фишера или критерия χ2 Пирсона с поправкой Йетса для нескольких групп.
Результаты
Исследование продолжалось с 24.10.2017 (дата первого стартового визита) по 10.12.2018 (дата завершения исследования последним участником).
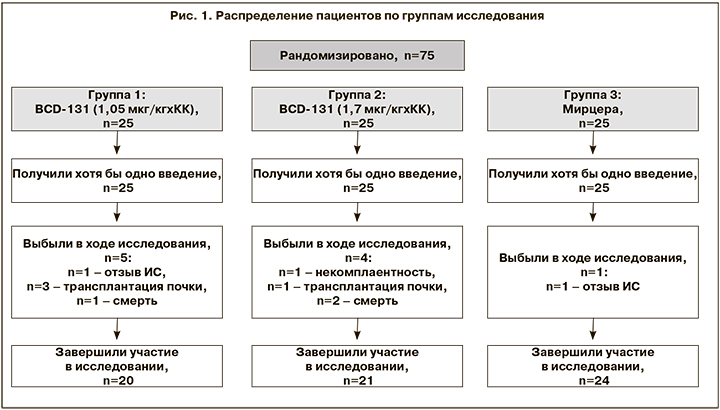
В общей сложности в исследование были рандомизированы 75 человек. В течение 21 недели терапии выбыли 10 пациентов. Причины выбывания: отзыв ИС, некомплаентность, трансплантация почки, летальный исход (не связанный с терапией). Таким образом, участие в исследовании завершили 65 пациентов: в группе 1 (BCD 1,05 мкг/кг) – n=20, в группе 2 (BCD 1,7 мкг/кг) – n=21, в группе 3 (Мирцера) – n=24 (рис. 1).
Оценка исходных демографических показателей не выявила значимых различий между группами по расовому составу (р=1,00, двусторонний точный критерий Фишера), соотношению мужчин и женщин (р=0,8472, критерий χ2 Пирсона с поправкой Йетса), росту (р=0,7387, критерий Краскела–Уоллиса), массе тела (р=0,9318, критерий Краскела–Уоллиса), индексу массы тела (р=0,8738, критерий Краскела–Уоллиса) и пр возрасту (р>0,05 для всех попарных сравнений, критерий Манна–Уитни с поправкой Беньямини–Йекутили).
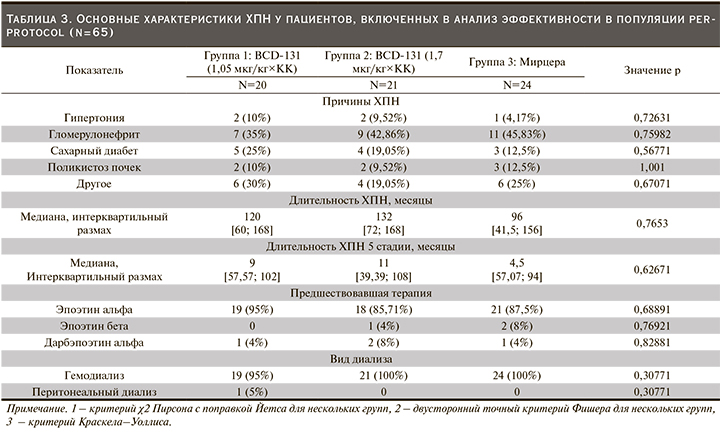
В основном включенным в исследование пациентов ХПН являлась исходом гломерулонефрита, сахарного диабета, реже причиной нарушения функции почек была гипертоническая болезнь. Продолжительность ХПН, а также длительность терминальной стадии заболевания не различались у пациентов всех трех групп. Абсолютное большинство участников до включения в исследование получали эпоэтин альфа: 95%, 86 и 88% пациентов первой, второй и третьей групп соответственно (р=0,6889, критерий χ2 Пирсона с поправкой Йетса) (табл. 3).
Число участников, имевших одно или несколько сопутствовавших заболеваний, было сопоставимым во всех группах. Наиболее часто встречались заболевания сердечно-сосудистой системы и желудочно-кишечного тракта. Артериальная гипертензия была диагностирована у 80%, 90,48 и 83,33% пациентов, ишемическая болезнь сердца – у 24%, 20 и 12% участников первой, второй и третьей групп соответственно (р=0,6024, двусторонний точный критерий Фишера для нескольких групп). Хронический гастрит или гастродуоденит наблюдался у 56%, 48 и 52% в группах 1, 2 и 3 соответственно (р=0, 8519, критерий χ2 Пирсона с поправкой Йетса для нескольких групп).
Анализ эффективности
На протяжении всего исследования cредний уровень гемоглобина находился в пределах целевых значений и составил 111,081±8,463 г/л, 113,796±8,242 и 113,472±9,152 г/л в группах BCD-131 1,05, BCD-131 1,7 мкг/кг и Мирцера соответственно (р=0,4147, критерий Манна–Уитни).
При сравнении среднего уровня гемоглобина, зарегистрированного у пациентов после завершения 21 недели терапии (среднее арифметическое уровня гемоглобина на неделях 21 и 23), с исходным уровнем гемоглобина (среднее арифметическое уровня гемоглобина на скрининге и на визите 1) не было выявлено статистически значимых различий между группами, несмотря на то что изменение показателя носило разнонаправленный характер (p=0,1059, критерий Краскелла–Уоллиса). В первой и третьей группах было отмечено снижение среднего уровня гемоглобина (-3,7±9,422 и -1,5±12,485 г/л соответственно), тогда как во второй группе наблюдалось увеличение среднего уровня гемоглобина на 3,786±12,729 г/л (представлены средние значения и стандартное отклонение). Однако такие колебания средних уровней гемоглобина были в рамках целевых значений во всех группах.
С целью подтверждения основной гипотезы исследования был рассчитан 95% ДИ для разницы средних арифметических значений показателя «изменение уровня гемоглобина в течение периода оценки по сравнению с исходным уровнем» между группой 1 исследуемого препарата (BCD-131 1,05 мкг/кг) и группой 3 (Мирцера), который составил [-8,87;4,47], что не превышает предустановленных границ эквивалентности 10,00 г/л. Таким образом, доказана эквивалентная эффективность исследуемого препарата BCD-131 в дозе 1,05 мкг/кг и препарата сравнения Мирцера. 95% ДИ, рассчитанный для разницы вышеуказанных показателей между группой 2 (BCD-131 1,7 мкг/кг) и группой 3 (Мирцера), составил [-2,32; 12,89], что выходит за пределы диапазона [-10,0; 10,0] г/л. Таким образом, гипотеза об эквивалентности препарата BCD-131 в дозе 1,7 мкг/кг и препарата сравнения Мирцера не подтверждена.
На протяжении исследования практически у всех пациентов на каком-либо из визитов отмечались отклонения уровня гемоглобина от целевых значений. Однако при сравнении трех групп относительное число пациентов с целевым уровнем гемоглобина (100–120 г/л) в течение недель 1–23 на всех визитах не различались (р>0,05, двусторонний точный критерий Фишера, при попарных сравнениях между группами – критерий Манна–Уитни с поправкой Беньямини–Йекутили). Обращало на себя внимание, что показатель «относительное число пациентов с целевым уровнем гемоглобина» был менее вариабельным в группе пациентов, получавших исследуемый препарат BCD-131 в дозе 1,05 мкг/кг.
Доза препаратов титровалась в зависимости от уровня гемоглобина для поддержания показателя в пределах 100–120 г/л, что соответствует актуальным рекомендациям по применению эритропоэз-стимулирующих препаратов [1]. Доля пациентов в группах, которым в ходе терапии потребовалась коррекция дозы исследуемого препарата или препарата сравнения, статистически не различалась и составила 60%, 52,38 и 66,67% пациентов первой, второй и третьей групп соответственно (p=0,6266, критерий χ2 Пирсона с поправкой Йетса).
Анализ безопасности
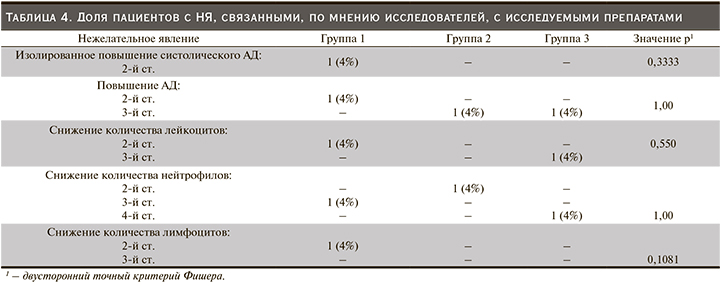
Препарат BCD-131 (обе исследованные дозы) и препарат сравнения Мирцера продемонстрировали сопоставимые профили безопасности на протяжении всего периода терапии: статистически значимых различий между тремя группами по количеству пациентов с зарегистрированными НЯ не обнаружено. В исследуемой популяции пациентов с терминальной стадией ХПН наиболее часто регистрировались повышение артериального давления (АД) 2–3-й ст. тяжести, изолированное повышение систолического или диастолического АД, снижение количества лимфоцитов 2–3-й ст., гипергликемия. По мнению исследователей, в большинстве случаев НЯ были обусловлены основным и/или сопутствовавшими заболеваниями. Доля пациентов, у которых была установлена связь НЯ с исследуемым препаратом, составила 20% в первой группе, 8% во второй и 8% в третьей (р=0,4861, двусторонний точный критерий Фишера) (табл. 4).
С клинической точки зрения наибольший интерес представляют НЯ 3–4-й ст. тяжести и СНЯ, а также оценка связи зарегистрированных НЯ и СНЯ с терапией.
Связь НЯ 3–4-й ст. тяжести с исследуемой терапией была установлена в единичных случаях. В группе препарата BCD-131 1,05 мкг/кг у одного пациента имело место снижение количества нейтрофилов 3-й ст., у второго – снижение количества лимфоцитов 3-й ст. Среди пациентов второй группы, получавших BCD-131 в дозе 1,7 мкг/кг, у одного участника было отмечено повышение АД 3-й ст. В третьей группе у одного пациента имело место повышение АД 3-й ст., у второго – снижение количества лейкоцитов 3-й ст. и снижение количества нейтрофилов 4-й ст.
Статистически значимых различий между группами не обнаружено (р=1,000, двусторонний точный критерий Фишера).
СНЯ были зарегистрированы у 24%, 16 и 12% участников первой, второй и третьей групп соответственно (р=0,6447, двусторонний точный критерий Фишера), во всех случаях стали следствием основного и/или сопутствовавших заболеваний и не были связаны с исследуемым препаратом/препаратом сравнения. СНЯ были представлены сердечно-сосудистыми и цереброваскулярными нарушениями, тромботическими осложнениями, инфекциями, ухудшением течения сахарного диабета.
Три участника досрочно прекратили участие в исследовании в связи с НЯ: группа 1 (n=1), группа 2 (n=2), р=0,7686 (двусторонний точный критерий Фишера). Во всех случаях причиной прекращения терапии оказалась смерть пациента, не связанная с применявшимися эритропоэз-стимулирующими препаратами. Причиной смерти пациента первой группы стал геморрагический инсульт. Во второй группе у одного пациента летальный исход наступил в результате острого инфаркта миокарда; у второго имел место инсулинозависимый сахарный диабет 2 типа, осложнившийся синдромом диабетической стопы, хронический остеомиелит, прогрессирующая хроническая сердечная недостаточность на фоне системной воспалительной реакции, атеросклеротический кардиосклероз.
Анализ частоты тромботических осложнений, в общей сложности наблюдавшихся у 6 из 75 пациентов, также не выявил статистически значимых различий между группами (р=1,00, двусторонний точный критерий Фишера). В первой группе у одного пациента был диагностирован геморрагический инсульт, у второго – острый инфаркт миокарда. Во второй группе у одного пациента был отмечен тромбоз сосудистого доступа, у второго диагностирован острый инфаркт миокарда. В третьей группе зафиксировано два случая тромбоза сосудистого доступа. Ни в одном из случаев не было отмечено связи тромботических осложнений с применением исследуемого препарата или препарата сравнения.
Анализ иммуногенности
Ни у одного из пациентов не было выявлено формирования САТ, в связи с чем анализ на наличие НАТ выполнен не был.
Обсуждение результатов
Обсуждение дизайна, исследуемой популяции и степени доказательности исследования
Препарат BCD-131 – новый оригинальный препарат, потенциальный терапевтический кандидат для лечения анемии у больных ХПН. Целью описанного международного многоцентрового рандомизированного открытого сравнительного клинического исследования II фазы стало установление терапевтически эффективной и безопасной дозы препарата BCD-131 при его многократном применении для лечения анемии у находившихся на диализе пациентов с ХПН.
Выбор препарата сравнения, а также дозы и кратности введения исследуемого препарата были основаны на данных, полученных в ходе клинического исследования I фазы, свидетельствовавших о том, что профиль фармакокинетики, фармакодинамики и безопасности препарата BCD-131 наиболее близок к соответствующим характеристикам препарата Мирцера. Режим введения препарата сравнения Мирцера соответствовал актуальной инструкции по медицинскому применению [12].
На первом этапе настоящего исследования были установлены дозы препарата BCD-131, обладающие наиболее благоприятным профилем фармакокинетики, фармакодинамики и безопасности. Целью второго этапа стала оценка и сравнение эффективности, безопасности и иммуногенности двух выбранных доз препарата BCD-131 (1,05 и 1,7 мкг/кг) и препарата Мирцера.
Одним из важных факторов, влияющих на достоверность результатов клинического исследования, является проведение качественной рандомизации, критериями которой признаны формирование случайной последовательности, определяющей распределение пациента в ту или иную группу терапии и сокрытие данной последовательности от исследователей [13].
В настоящем исследовании оба условия были соблюдены. Метод блочной рандомизации позволил избежать предвзятости и субъективной оценки результатов лечения. Проводившаяся перед рандомизацией стратификация участников обеспечила максимальную равноценность групп. С учетом описанных выше особенностей дизайна исследования заслепление терапии не проводилось, что можно расценить как одно из ограничений данного исследования. Однако, принимая во внимание, что оценка эффективности терапии проводилась по объективному лабораторному показателю «изменение уровня гемоглобина в течение периода оценки», являющегося одним из наиболее чувствительных параметров при оценке эффективности и определении эквивалентности свойств эритропоэз-стимулирующих препаратов, проведение открытого исследования представляется приемлемым и целесообразным.
Таким образом, дизайн представленного исследования, объем выборки, общая продолжительность периода применения препаратов, исследуемая популяция, первичная конечная точка для оценки эффективности соответствуют международным стандартам и обусловливают высокую степень доказательности данного исследования II фазы [14].
Обсуждение полученных данных по эффективности, безопасности и иммуногенности
В рамках настоящего исследования на протяжении 21 недели терапии средний уровень гемоглобина у пациентов, получавших как исследуемый препарат BCD-131 (обе исследованные дозы), так и препарат сравнения Мирцера, находился в пределах целевого диапазона. Показатель «изменение уровня гемоглобина», выбранный в качестве первичной конечной точки оценки эффективности, стал основным критерием, использованным для доказательства гипотезы эквивалентности свойств исследуемого препарата и препарата сравнения. Проведенный анализ позволил констатировать эквивалентную эффективность препарата BCD-131 в дозе 1,05 мкг/кг и Мирцера. Также не было обнаружено статистически значимых различий между группами по всем прочим оцененным параметрам эффективности. Таким образом, показано, что препарат BCD-131 позволяет поддерживать концентрацию гемоглобина, необходимую для контроля симптомов анемии.
Каких-либо новых данных по безопасности эритропоэз-стимулирующих препаратов в ходе настоящего исследования II фазы не получено. В целом продемонстрированы благоприятный профиль безопасности и удовлетворительная переносимость как исследуемого препарата (в обеих дозах), так и препарата сравнения.
Закономерно, что в популяции пациентов с терминальной стадией ХПН, имевших одно или несколько сопутствовавших заболеваний, наблюдалась высокая частота НЯ/СНЯ, которая в абсолютном большинстве случаев не имела причинно-следственной связи с исследуемой терапией, а служила закономерными проявлениеми (как клиническим, так и лабораторным) основного и многочисленных сопутствовавших заболеваний. Среди НЯ, связанных, по мнению исследователей, с терапией, наиболее часто регистрировалось повышение АД. Данные литературы свидетельствуют о том, что применение эритропоэзстимулирующих препаратов может быть ассоциировано как с усугублением имеющейся артериальной гипертензии, так и с ее развитием de novo на фоне терапии.
В основе развития эритропоэтин-ассоциированной артериальной гипертензии лежит нарушение баланса между процессами вазодилатации и вазоконстрикции. Показано, что важную роль в патогенезе повышения АД в таких случаях играют простаноиды, эндотелин, оксид азота, а также ассоциированное с введением эритропоэтина повышение чувствительности к катехоламинам и ангиотензину II [15].
Ряд зарегистрированных у пациентов лабораторных отклонений (лимфо-, лейко- и нейтропения) были расценены исследователями как связанные с терапией. Ассоциация хронической болезни почек с иммунной дисфункцией, в том числе сопровождающейся снижением числа лимфоцитов, хорошо известна как ученым, так и практикующим нефрологам. В одном из исследований, посвященных этой проблеме и включившем 192 пациента с ХПН, лимфопения наблюдалась у половины участников. Патогенез возникновения данных нарушений в настоящее время продолжает изучаться [16, 17]. Важно учитывать, что в рамках данного клинического исследования применялись критерии ВОЗ для оценки связи НЯ с исследуемым продуктом, согласно которым НЯ должно быть признано связанным с применяемой терапией в случаях, когда развитие НЯ по времени связано с приемом препарата, даже если это можно объяснить наличием сопутствующих заболеваний или приемом других лекарственных препаратов и влиянием химических соединений [18]. Таким образом, вероятно, указанные лабораторные отклонения обусловлены основным заболеванием и патогенетически не связаны с введением препаратов эритропоэтина, однако в соответствии с международными стандартами в условиях клинического исследования они должны быть расценены как связанные с исследуемыми препаратами.
Ни у одного из пациентов не было обнаружено связывающих антител к пегилированному дарбэпоэтину/метоксиполиэтиленгликоль-эпоэтину бета, что свидетельствует о низкой иммуногенности исследуемого препарата и препарата сравнения.
Заключение
BCD-131 представляет собой инновационный оригинальный препарат пегилированного дарбэпоэтина. В рамках доклинических исследований были детально изучены структура молекулы, ее физико-химические свойства, специфическая активность in vitro и in vivo. Результаты доклинических исследований позволили сделать вывод об отсутствии значимого токсического влияния препарата на общее состояние, а также на органы и системы экспериментальных животных.
В исследовании I фазы показано, что профиль фармакокинетики, фармакодинамики и безопасности исследуемого препарата BCD-131 наиболее близок к соответствующим показателям препарата Мирцера.
В рамках представленного исследования II фазы, проведенного в популяции пациентов с терминальной стадией ХПН, установлено, что оптимальной терапевтической дозой BCD-131 является доза 1,05 мкг/кг. Подтверждена гипотеза об эквивалентной эффективности исследуемого препарата BCD-131 в дозе 1,05 мкг/кг и препарата Мирцера. Анализ безопасности продемонстрировал отсутствие статистически значимых различий в частоте регистрации НЯ/СНЯ, в том числе НЯ 3–4-й ст. тяжести, и событий, связанных, по мнению исследователей, с терапией.


