
Инсулинорезистентность (ИР) определяется как снижение биологического действия инсулина на его органы-мишени (прежде всего печень и скелетные мышцы) [1]. В основе ее лежит нарушение передачи сигналов инсулина на рецепторном или пострецепторном уровнях, проявляющаяся в нарушении захвата, метаболизма или накопления глюкозы в мышечной, печеночной и жировой тканях [2].
Клиническим проявлением ИР служат нарушение толерантности к глюкозе, гиперинсулинемия, гипергликемия и дислипидемия. ИР может быть как физиологической (например, при беременности), так и патологической (например, при сахарном диабете 2 типа – СД2), как первичной (например, при СД2) так и вторичной, часто сопровождающей заболевания обмена веществ (например, акромегалия, синдром Кушинга, феохромоцитома, гипертиреоз) [3, 4, 132, 163].
Чувствительность тканей к инсулину может изменяться под воздействием таких факторов, как масса тела, количество висцерального жира и концентрация свободных жирных кислот и глюкокортикоидов в крови [5, 6]. Инсулин подавляет печеночный глюконеогенез и гликолиз, ингибирует липолиз в жировой ткани и стимулирует поглощение глюкозы скелетными мышцами [1]. При ИР в условиях снижения чувствительности к инсулину и необходимости поддержания физиологического уровня глюкозы крови синтез и секреция инсулина β-клетками островков Лангерганса поджелудочной железы увеличиваются, что ведет к хронической гиперинсулинемии [7].
По мере прогрессирования ИР дальнейшего повышения секреции инсулина поджелудочной железой становится недостаточно для поддержания нормогликемии, что ведет к клиническим проявлениям СД2 [8], причем хроническая гипергликемия подавляет экспрессию рецепторов инсулина в скелетных мышцах, печеночной и жировой тканях, что еще больше снижает их чувствительность к инсулину [9–14].
Методы оценки ИР
В настоящее время в клинической и исследовательской практике существует множество методов количественной оценки ИР различной степени сложности, инвазивности и трудоемкости [1].
Динамические модели основаны на пероральном или парентеральном введении глюкозы или глюкозы с инсулином в организм и в первую очередь отражают периферическую ИР (т.е. утилизацию глюкозы скелетных мышцах), тогда как статические модели основаны на определении глюкозы и инсулина крови натощак, фокусируясь в основном на печеночном глюконеогенезе [2, 7, 16]. Динамические модели обеспечивают более точную оценку чувствительности к инсулину по сравнению со статическими моделями, однако отнимают значительно больше времени и ресурсов и поэтому редко применяются в эпидемиологических исследованиях [2]. К группе динамических моделей оценки ИР относятся пероральный глюкозотолерантный тест (ПГТТ), наиболее простой, динамический и часто применяемый в клинической практике тест [17, 18], и внутривенный глюкозотолерантный тест, применяемый в основном в клинических исследованиях. Оба метода основаны на изменениях концентраций глюкозы и инсулина в крови в ответ на пероральную или внутривенную стандартную нагрузку глюкозой [19].
Другим динамическим является метод эугликемического гиперинсулинемического клэмпа (ЭГК), служащий на протяжении более чем 40 лет эталоном прямого количественного определения инсулинообусловленнoго поглощения глюкозы скелетными мышцами [20]. Для определения ИР этим методом требуется поддержание эугликемии на фоне подавления печеночного глюконеогенеза путем создания высокого уровня инсулина в плазме, что достигается непрерывной инфузией инсулина и глюкозы [20]. По достижении устойчивого уровня эугликемии (при стабильной концентрации инсулина в крови) количество вводимой глюкозы соответствует скорости ее захвата тканями, т.е. отражает инсулинообусловленный метаболизм глюкозы в тканях. При наличии резистентности к инсулину для поддержания эугликемии требуются меньшие количества глюкозы. Таким образом, скорость введения глюкозы представляет собой прямую меру чувствительности организма к инсулину [1, 8].
Хотя ЭГК и является самым точным и надежным методом оценки ИР, позволяющим определять степень поглощения глюкозы скелетными мышцами, он применяется в основном при проведении фармакодинамических клинических испытаний, например при разработке антидиабетических лекарственных средств, и практически не используется в клинической практике ввиду своей трудоемкости, инвазивности и необходимости дополнительного технического оснащения и специально обученного персонала [1, 8, 23]. Вместо него для оценки ИР обычно применяют недорогие и практичные косвенные статические тесты, использующие оценку чувствительности к инсулину по концентрации глюкозы и инсулина натощак и не требующие больших затрат времени [8].
Одним из таких тестов является наиболее часто используемый индекс HOMA-IR (Homeostasis Model Assessment of Insulin Resistance), который широко применяется в эпидемиологических исследованиях, при которых не требуется высокой точности оценки чувствительности к инсулину [24]. Результаты оценки ИР этим методом положительно коррелируют с концентрацией глюкозы или инсулина крови натощак и рассчитываются по формуле: HOMA-IR=уровень инсулина натощак (мкЕД/мл), уровень глюкозы натощак (ммоль/л)/22 [3, 26]. Другим популярным тестом является количественный индекс оценки чувствительности к инсулину QUICKI (Quantitative Insulin Sensitivity Check Index), который рассчитывается по формуле QUICKI=1/[log(I0)+log(G0)], где I0 – уровень инсулина натощак (мкЕД/мл) и G0 – уровень глюкозы натощак (мг/дл) [27].
Важно отметить, что у людей с нормальной функцией почек результаты как пероральной динамической ПГТТ, так и статической модели HOMA-IR достаточно хорошо коррелируют с таковыми, получаемыми посредством ЭГК, являющегося «золотым» стандартом оценки ИР (коэффициенты корреляции R=0,7–0,9) [8].
Однако у пациентов со снижением функции почек корреляция между результатами статических индексов ИР (HOMA-IR, QUICKI) и результатами ЭГК выражена слабее по сравнению с корреляцией между результатами динамических тестов (ПГТТ) и ЭГК [2], т.к. при хронической болезни почек (ХБП) снижение почечного клиренса инсулина может повышать концентрацию инсулина натощак и опосредованно оказывать влияние на концентрацию глюкозы натощак [28], являющихся детерминантами статической оценки ИР в моделях HOMA-IR и QUICKI [8].
Более того, при интерпретации результатов статических тестов следует делать поправку на следующее: у больных ХБП основной вклад в нарушение чувствительности к инсулину играют скелетные мышцы, а концентрация глюкозы крови натощак, которая, как упомянуто выше, служит основным параметром статических тестов, в первую очередь определяется не снижением действия инсулина в мышечной ткани, а ночным печеночным глюконеогенезом [8]. Кроме этого важно также отметить, что у больных в терминальной стадии ХБП заместительная почечная терапия (перитонеальный диализ или гемодиализ) может оказывать непосредственное влияние на уровень гликемии, тем самым и на точность определения ИР методом статических тестов [23].
Оценка влияния почечной функции на результаты тестов оценки ИР дана в исследовании T. Jia et al. [28]. Авторы сравнили результаты оценки ИР, основанные на методах HOMA-IR или ПГТТ, с таковыми ЭГК у пожилых, не страдающих СД людей, которые были стратифицированы по скорости клубочковой фильтрации (СКФ): <60 мл/мин/1,73 м2 (n=495) и ≥60 мл/мин/1,73 м2 (n=579). Оказалось, что результаты оценки ИР у участников с СКФ<60 мл/мин/1,73 м2, полученные с помощью метода HOMA-IR слабее, хотя и не статистически значимо коррелировали с результатами ЭГК, чем у участников с более сохранной почечной функцией (RСКФ<60=0,67 vs. RСКФ>60 =0,71) [14].
В то же время точность оценки ИР на основе ПГTT при использовании Matsuda index, который рассчитывается по формуле: ISIMatsuda=10, 000/√G0×I0×Gmean×Imean, где G0 (мг/дл) и I0 (мкЕД/мл), концентрация глюкозы и инсулина в плазме крови натощак, а Gmean и Imean средняя концентрация глюкозы (мг/дл) и инсулина (мкЕд/мл) в плазме крови во время ПГТТ (0–120 минут) у участников с СКФ<60 мл/ мин/1,73 м2 превышала точность оценки ИР методом HOMAIR (RПГTT=0,74 vs RHOMA-IR=0,67) [8].
В совокупности эти данные свидетельствуют: при нарушении почечной функции как статические (например, HOMA-IR), так и динамические модели (например, ПГTT) гарантируют достаточную точность оценки ИР в клинической практике, хотя модель ПГТТ несколько превосходит статические модели в достоверности оценки [8].
ХБП и ИР
Клинические исследования последних лет показали, что ИР может проявляться на разных стадиях снижения почечной функции без взаимосвязи с избыточной массой тела [30–33, 133–136, 161], причем степень выраженности ИР положительно коррелирует со стадией ХБП даже в отсутствие СД [34].
Тот факт, что синдром резистентности к инсулину может возникать у лиц с заболеваниями почек различной этиологии (например, аутосомно-доминантным поликистозом почек или IgA-нефропатией) еще до существенного снижения СКФ указывает на то, что ни этиология нефропатии, ни наличие поздних проявлений/осложнений ХБП (уремия, почечная анемия, вторичный гиперпаратиреоз, недостаточность витамина D или метаболический ацидоз) не могут рассматриваться как определяющие факторы возникновения ИР у больных ХБП [1, 35].
ХБП и гомеостаз глюкозы
Продукция и утилизация глюкозы почками – важные компоненты метаболизма глюкозы в организме человека [36, 37]. Поэтому нарушение функции почек само по себе может служить одной из причин ИР [1].
С другой стороны, при ХБП, особенно на ее поздних стадиях, снижение чувствительности к инсулину преимущественно обусловлено воздействием уремических токсинов на пост-рецепторную передачу сигнала инсулина в мышечных клетках [34, 38], приводящее к снижению утилизации глюкозы скелетными мышцами. При этом утилизация глюкозы клетками печени и ингибирующее действие инсулина на печеночный глюконеогенез также могут снижаться, хотя и в меньшей степени [39–42].

Известно, что по мере прогрессирования ХБП у большинства больных СД гликемический контроль спонтанно улучшается [43]. В поздних стадиях ХБП риск выраженной гипогликемии (<70 г/дл) существенно возрастает, причем вероятность развития тяжелой гипогликемии может увеличиваться в 5 раз [44], делая необходимым снижение дозировки или полное прекращение терапии инсулином или другими антигликемическими препаратами [43, 46–59]. В терминальной стадии ХБП суточная доза инсулина может снижаться на 28–60%, а примерно в 30% случаев у ранее инсулинозависимых больных СД инсулин вообще не требуется [23, 50, 51]. Такое снижение в его потребности во многом объясняется снижением почечного и печеночного клиренса инсулина [53, 54], почечного глюконеогенеза у пациентов с терминальной ХБП, гипогликемическим эффектом гемодиализа, особенно при использовании диализата с низким или нормальным уровнем глюкозы [49, 56, 57].
Причины и механизм ИР при ХБП
Причины ИР при ХБП многофакторны: малоподвижный образ жизни, нездоровое питание, курение, накопление висцерального жира, измененная кишечная флора, дизрегуляция секреции адипокинов со снижением уровня адипонектина, наличие статуса хронического системного воспаления, повышенная продукция активных форм кислорода, ответственных за окислительный стресс, активация ренин-ангиотензин-альдостероновой системы (РААС), эндотелиальная дисфункция, метаболический ацидоз, дефицит витамина D, почечная анемия и уремические токсины (см. рисунок) [58, 59]. Более того, СД и ожирение значительно усугубляют проявления ИР при ХБП [7, 23, 60, 63–65, 67, 68, 71, 140, 141, 147, 148, 162, 164].
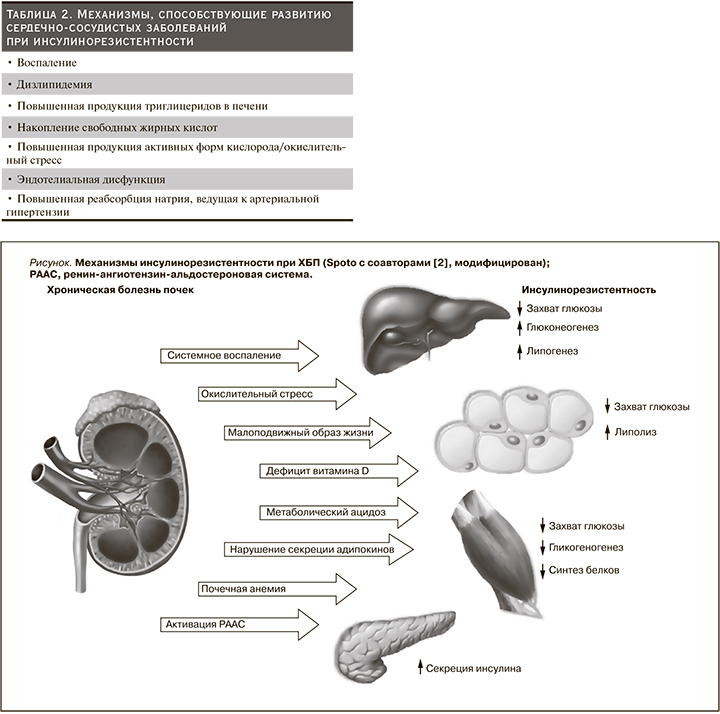
В настоящее время доказано участие прои противовоспалительных цитокинов в механизме ИР у больных с нарушением функции почек:
1) ИР и ожирение связаны с повышением экспрессии фактора некроза опухоли α (ФНО-α, TNF-α) в жировой и мышечной тканях [69];
2) ИР ассоциирована с повышением уровня интерлейкина1β (ИЛ-1β), который снижает чувствительность β-клеток островков Лангерганса поджелудочной железы к инсулину и приводит к нарушению секреции и передачи сигналов инсулина в периферических тканях и макрофагах [70];
3) повышение уровня ИЛ-6 способствует развитию ИР, снижая экспрессию субстрата рецептора инсулина-1 (IRS1) и транспортера глюкозы-4 (GLUT-4) [71, 72] и повышая концентрацию триглицеридов в крови [73];
4) резистин, вырабатываемый мононуклеарными клетками периферической крови, принимает участие в патогенезе ИР, ожирения и СД2 [74];
5) лептин снижает концентрацию глюкозы крови, ингибируя глюконеогенез, и улучшает чувствительность тканей к инсулину, препятствуя накоплению триглицеридов в печени и скелетных мышцах [75–77]. Резистентность к лептину, обычно наблюдающейся при ожирении, ассоциирована с ИР и нарушением секреции инсулина, в т.ч. вследствие накопления триглицеридов в тканях-мишенях [63, 137];
6) ингибитор активатора плазминогена-1 (PAI-1), являющийся провоспалительным адипокином, играет важную роль в развитии ИР даже в отсутствие ожирения [79, 80];
7) повышение моноцитарного хемоаттрактантного протеина-1 (MCP-1), вызывающего дедифференцировку адипоцитов, способствует развитию ИР, ожирения и СД2 [25];
8) адипонектин, являющийся единственным противовоспалительным цитокином, повышает чувствительность тканей к инсулину [82–84, 137, 139, 143].
При этом важно отметить, что степень нарушения почечной функции отрицательно коррелирует с выраженностью статуса хронического системного воспаления, уровнем секреции провоспалительных цитокинов (например, ИЛ-1β, -6, ФНО-α) и некоторых провоспалительных протеинов (С-реактивного белка и фибриногена), играющих ключевую роль в развитии ИР [85].
Другим важным фактором, играющим роль в снижении чувствительности к инсулину при ХБП, является активация РААС, приводящая к увеличению продукции асимметричного диметиларгинина (АДМА) в адипоцитах и стимуляции экспрессии ИЛ-6 [86, 87]. Это способствует нарушению функции рецептора инсулина (IRS-1) с нарушением передачи инсулинообусловленнoго сигнала внутри клетки и последующим снижением функции GLUT-4, что в итоге ведет к нарушению захвата глюкозы мышечной и жировой тканями и развитию ИР [88]. При этом, например, показано, что блокада рецепторов ангиотензина II олмесартаном у пациентов с нарушенной функцией почек может оказывать позитивное воздействие на резистентность к инсулину [89].
Следующим фактором, способствующим развитию ИР, является метаболический ацидоз, который вызывает снижение аффинитета рецептора инсулина и нарушению инсулинообусловленной передачи сигналов в клетках органов-мишеней даже в начальных стадиях ХБП [90–92].
Механизм влияния ИР на ХБП
Инсулин играет важную роль в регуляции почечного кровотока и клубочковой фильтрации. В условиях резистентности тканей к инсулину биодоступность оксида азота (NO) снижается, что приводит к нарушению NO-зависимой релаксации почечных сосудов, внутрисосудистому рекрутированию лейкоцитов и моноцитов, агрегации и адгезии тромбоцитов, усилению выраженности статуса хронического воспаления и в итоге запускает механизмы, способствующие развитию почечного фиброза. Кроме этого снижение продукции NO вызывает нарушение канальцево-гломерулярной обратной связи – механизма, с помощью которого осуществляется ауторегуляция почечного кровотока и клубочковой фильтрации, что в свою очередь приводит к гиперфильтрации и задержке натрия в организме и может повышать риск развития артериальной гипертензии [126–131, 138, 142, 162, 166].
Меры воздействия на ИР
К мерам воздействия на ИР, которые улучшают клинический прогноз у пациентов с ХБП, относятся модификация образа жизни, лекарственная терапия (например, метформин, тиазолидиндионы, агонисты рецепторов глюкагоноподобного пептида-1 и ингибиторы натрий-глюкозного котранспортера 2) и заместительная почечная терапия.
Модификация образа жизни. Одним из самых эффективных методов лечения ИР, имеющим положительный эффект на развитие сердечно-сосудистых осложнений и продолжительность жизни больных ХБП, является изменение образа жизни включающее увеличение физической активности, отказ от курения и изменение диеты [147]. Показано, например, что увеличение физической активности повышает чувствительность к инсулину при ожирении [94], а низкобелковая диета, дополненная незаменимыми аминокислотами и кетоаналогами, уменьшая продукцию азотистых соединений, улучшает чувствительность к инсулину пациентов в поздних стадиях ХБП [45, 93].
Лекарственные средства. Метформин является сенсибилизатором инсулина и одним из наиболее часто используемых препаратов для лечения ИР при СД2, главным органом-мишенью которого является печень, где он угнетает глюконеогенез. Терапия метформином снижает массу тела [96] и уменьшает риск возникновения СД у людей с гипергликемией натощак или постпрандиальной гипергликемией [93, 98, 145]. Применение метформина пациентами на ранних стадиях ХБП ассоциировано с существенным – на 33% – снижением смертности [99]. Однако важно помнить, что т.к. метформин выводится из организма в неизмененном виде практически исключительно почками, его применение на поздних стадиях ХБП (СКФ<30 мл/мин) противопоказано по причине повышенного риска его накопления в организме и развития лактоацидоза.
Агонисты рецепторов глюкагоноподобного пептида-1, или миметики инкретина, усиливают глукозоопосредованную секрецию инсулина, замедляют опорожнение желудка и снижают постпрандиальный глюкагон и потребление пищи. Известно, например, что представитель этой группы лираглутид улучшает прогноз заболеваний сердечно-сосудистой системы и почек у пациентов с СД [100]. Однако, хотя и известно, что агонисты рецепторов глюкагоноподобного пептида-1 положительно влияют на чувствительность к инсулину в экспериментах на животных [101], их влияние на ИР и на риск возникновения сердечно-сосудистых и почечных осложнений в отсутствие СД еще требует подтверждения [144, 149–154, 157, 158].
Ингибиторы натрий-глюкозного котранспортера 2 (SGLT2i) представляют собой класс пероральных антидиабетических лекарственных средств, механизм действия которых связан со снижением реабсорбции глюкозы в почках. Показано, что такие ингибиторы SGLT2, как эмпаглифлозин и канаглифлозин, замедляют прогрессирование ХБП и улучшают прогноз сердечно-сосудистые заболеваний у пациентов с заболеваниями почек как с СД, так и в его отсутствие [19, 102, 105, 155, 156, 159, 160]. Кроме того, лекарственные препараты этой группы способны повышать чувствительность к инсулину у больных СД [106, 107, 145, 146], что происходит, скорее всего, вследствие уменьшения массы тела, обусловленного потерей калорий в виде почечной экскреции глюкозы, хотя еще нет достаточной доказательной базы их влияния на чувствительность к инсулину у больных ХБП в отсутствие СД.
У пациентов с ХБП и СД тиазолидиндионы (TZD) улучшают чувствительность к инсулину посредством активации PPAR-γ, повышения уровня адипонектина [58, 108–110], нормализации липидного профиля [111] и уменьшения хронического системного воспаления. В отношении улучшения клинического прогноза известно, что, например, пиоглитазон препятствует развитию микрои макрососудистых осложнений, уменьшает вероятность повторного инсульта и острого коронарного синдрома у пациентов с ИР даже в отсутствие СД [112, 113]. Конечно, с учетом их побочных эффектов (увеличение массы тела, задержка жидкости, повышенный риск застойной сердечной недостаточности и остеопороза и, хотя и очень редко, гепатотоксичность и рак мочевого пузыря) [15, 23, 123–125], TZD должны применяться с осторожностью при прогрессирующей ХБП [114–122].
Клинические исследования показали, что ингибиторы ангиотензинпревращающего фермента и блокаторы рецепторов ангиотензина II способны повышать чувствительность к инсулину и снижать его концентрацию в крови больных артериальной гипертензией и ХБП, включая ее терминальную стадию (например, телмисартан у больных на перитонеальном диализе). Механизм их действия включает, кроме всего прочего, противодействие окислительному стрессу и улучшение микроциркуляции в мышцах, что нормализует передачу инсулиноопосредованного сигнала на пострецепторном уровне и повышает доступность глюкозы и инсулина в тканях-мишенях, прежде всего, в миоцитах [78, 81, 91].
Меры воздействия на ИР в поздних стадиях ХБП включают коррекцию ацидоза, почечной анемии, дефицита витамина D, вторичного гиперпаратиреоидизма и заместительную почечную терапию.
Например, известно, что коррекция ацидоза бикарбонатами и диета с повышенным содержанием овощей и фруктов улучшает чувствительность к инсулину [21, 29, 55, 61], а лечение почечной анемии эритропоэтином уменьшает выраженность ИР у пациентов, находящихся на диализе. При этом позитивный эффект связан не только с непосредственным влиянием эритропоэтина на ткани-мишени, но и со снижением мышечного катаболизма и повышением физической активности в связи с улучшением аэробных способностей мышечной ткани [2, 22, 62, 95, 103, 104, 119].
В последнее время удалось показать, что ИР может наблюдаться при дефиците витамина D, а терапия витамином D у больных ХБП ассоциирована с улучшением ИР [165]. Хотя механизмы, лежащие в основе положительного влияния витамина D на чувствительность к инсулину в поздних стадиях ХБП, не до конца понятны, известно, что, например, повышенная концентрация паратиреоидного гормона (ПТГ) ингибирует секрецию инсулина, в то время как коррекция вторичного гиперпаратиреоидизма путем паратиреоидэктомии нормализует ее [39, 52, 66]. С другой стороны, также известно, что у пациентов с терминальной ХБП, находящихся на гемодиализе, внутривенные активированные формы витамина D могут повышать инсулиночувствительность, не оказывая влияния на концентрацию ПТГ в крови [52].
Заместительная почечная терапия. Хорошо известно, что как гемо-, так и перитонеальный диализ улучшают чувствительность к инсулину пациентов в терминальной стадии ХБП [34, 45].
Заключение
ИР может сопровождать ХБП вне зависимости от наличия ожирения или СД и являться как служить следствием дисфункции почек, так и ускорять течение почечного заболевания.
Причины развития ИР у пациентов с ХБП включают как особенности образа жизни (например, гиподинамия, нездоровое питание), так проявление хронического нарушения функции почек (например, метаболический ацидоз, дефицит витамина D, почечная анемия и уремический статус), причем ожирение и СД дополнительно усугубляют ее течение.
Механизм развития ИР при нарушении функции почек включает хроническое системное воспаление, окислительный стресс, дизрегуляцию секреции адипокинов и активацию РААС. Недорогие и практичные динамические (например, пероральный глюкозотолерантный тест, основанный на изменениях концентраций глюкозы и инсулина в крови в ответ на пероральную стандартную нагрузку глюкозой) и статические (например, индекс инсулинорезистентности HOMA-IR, использующий оценку чувствительности к инсулину по концентрации глюкозы и инсулина натощак) модели оценки ИР дают достаточно точную количественную оценку чувствительности к инсулину у пациентов с ХБП.
Так как ИР повышает риск как сердечно-сосудистой заболеваемости и смертности, так и развития сердечно-сосудистых и почечных осложнений у больных ХБП, необходимо своевременное принятие эффективных мер профилактики и лечения этого состояния, включающих модификацию образа жизни, целенаправленную фармакотерапию, а в терминальной стадии заболевания почек – заместительную почечную терапию.


