
Введение
БФ связана с мутациями в гене GLA, кодирующем фермент α-галактозидазу А (α-Гал A), что приводит к снижению или отсутствию активности данного фермента и накоплению гликосфинголипидов в лизосомах разных клеток (эндотелиальных и гладкомышечных клетках сосудов, центральной нервной системы, сердца, эпителиальных клетках большинства органов). Это вызывает поражение сердца, почек, головного мозга, кожи и нервной системы. Различают классическую форму БФ с мультисистемной патологией и атипичную с поражением одной системы органов. Основным накапливаемым субстратом при БФ являются глоботриаозилцерамид (Gb3), а также его деацилированная форма – глоботриаозилсфингозин (LysoGb3).
БФ относится к редким заболеваниям. Ее частота в различных странах варьируется в широких пределах (от 1:40 000 до 1:117 000) [1]. Однако есть данные о высокой частоте БФ при проведении неонатального скрининга: в Италии частота составила 1:3100 живых новорожденных мальчиков [2], в Тайване – 1:1250 [3]. Довольно интересно, что большинство выявленных случаев относится к атипичной «кардиологической» форме БФ с пиком манифестации на 4–5-м десятилетиях жизни.
К основным симптомам классической формы БФ на развернутой стадии болезни относятся акропарестезии, ангиокератомы, почечная недостаточность, гипертрофия левого желудочка, а также гипогидроз, головные боли, нарушение слуха, желудочно-кишечные расстройства, быстрая утомляемость и «катаракта Фабри» [4].
Накопление гликосфинголипидов при БФ в подоцитах, эндотелиальных, эпителиальных и других клетках почек приводит к развитию почечных нарушений, проявляющихся протеинурией и снижением скорости клубочковой фильтрации (СКФ), ведущих в конечном итоге к развитию гломерулосклероза, тубулярной атрофии и ремоделирующих почечные ткани фиброзом, с возникновением на поздних стадиях терминальной почечной недостаточности [5]. У пациентов, не получающих специфического лечения, развивается постепенно прогрессирующее поражение почек, при котором можно выделить несколько стадий [6]. Первые проявления нефропатии появляются уже в раннем детском или подростковом возрасте в виде клубочковой гиперфильтрации. Второй клинический этап характеризуется поражением почек с нарушением концентрационной функции почек, развитием микроальбуминурии, протеинурии, появлением липидов в моче, с выделением кристаллов, которое можно обнаружить при проведении микроскопии осадка мочи. На третьем этапе отмечается прогрессирование нефропатии с развитием тяжелого поражения почек, а также присоединение сердечно-сосудистых и цереброваскулярных осложнений. Терминальная стадия хронической болезни почек (ТХБП) – частое проявление БФ у лиц мужского пола, наблюдаемое на 3–5-м десятилетиях жизни [7].
При изолированном поражении почек диагноз БФ нередко остается нераспознанным вплоть до необратимого ухудшения их фильтрационной функции, в связи с чем скрининговое обследование, предпринимаемое для обнаружения этого заболевания у пациентов с ТХПН, оказывается успешным. В 2013 г. консенсус экспертов рекомендовал проведение скрининга на наличие БФ у мужчин с признаками хронической болезни почек (ХБП) в возрасте до 50 лет, у которых отсутствует подтвержденный диагноз заболевания почек, и у женщин с ХБП неуточненного генеза вне зависимости от их возраста или с наличием других типичных клинических симптомов, подозрительных в отношении наличия БФ [8]. Применяемые методы скрининга: ферментная диагностика – измерение активности α-Гал A в пятнах высушенной крови мужчин с последующим молекулярно-генетическим подтверждением мутаций гена при положительном результате ферментного теста и молекулярно-генетическое тестирование в качестве основного лабораторного теста при проведении скрининга у женщин. Разница в методах и алгоритмах скринирования мужчин и женщин связана с тем, что α-Гал A находится в пределах референсных значений более чем у 60% женщин – носительниц БФ, у больных мужчин активность его всегда снижена [9]. LysoGb3 показывает бóльшую чувствительность, а измерение соотношения α-Гал A/LysoGb3, как было показано нами сравнительно недавно, более эффективно для выявления женщин с БФ [10].
В данной работе представлены результаты селективного скрининга пациентов с патологией почек с применением метода определения концентрации LysoGb3 в сухих пятнах крови с последующим подтверждающим ДНК-анализом.
Материал и методы
Образцы крови. Забор образцов капиллярной крови осуществлялся на стандартную фильтр-карточку Whatman 903 (Whatman GmbH, Germany). Образцы собирали до проведения гемодиализа и после высушивания при комнатной температуре, хранили до проведения исследования при t 4°C.
Перед взятием образцов крови все пациенты дали письменное информированное согласие на проведение анализа.
Измерение концентрации LysoGb3. Определение концентрации LysoGb3 проводилось в сухих пятнах крови с использованием метода высокоэффективной жидкостной хроматографии тандемной масс-спектрометрии, с помощью Nexera MS-MS ABI SCIEX 5500 QTrap и было описано ранее [11].
Измерение активности α-Гал A. Активность α-Гал A измеряли с помощью набора NeoLSD™ методом тандемной масс-спектрометрии с ионизацией в электроспрее на приборе AB SCIEX 3200 Qtrap [12].
Молекулярно-генетический анализ. Геномную ДНК выделяли из образцов сухих пятен крови с использованием набора для очистки геномной ДНК GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific). Экзоны 1–7 GLA и смежные интронные последовательности анализировали секвенированием по Сэнгеру на генетическом анализаторе ABI PRISM 3500xL (Applied Biosystems, США). Последовательность праймеров может быть представлена по запросу.
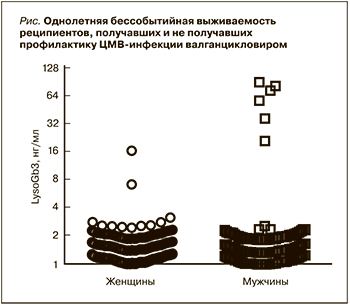 Результаты
Результаты
Данное исследование проводилось с 2017 по 2019 г. В него были включены 4078 пациентов: 2454 женщины (57,16±15,37 года) и 1623 мужчины (54,90±15,18 года) из отделений гемодиализа и нефрологии. Были выявлены три женщины с повышенным уровнем LysoGb3 (cut-off≥3), генетический анализ показал наличие мутации у одной из них. Биоматериал для проведения расширенного молекулярно-генетического исследования двух других пациенток предоставлен не был. У 6 мужчин выявлено повышение концентрации LysoGb3 (см. рисунок, график представлен двоичным логарифмом). Снижение активности фермента и патогенные мутации обнаружены у всех пациентов мужского пола. Распространенность БФ в данной выборке составила 0,17%.
Проведение семейного скрининга (определение концентрации LysoGb3, определение активности α-Гал A, поиск семейных мутаций) среди родственников выявленных пациентов позволило выявить дополнительно 5 пациентов (1 мужчина и 4 женщины). Данные по концентрации LysoGb3, активности фермента и соотношения α-Гал A/LysoGb3, а также результаты ДНК-диагностики выявленных пациентов приведены в табл. 1.
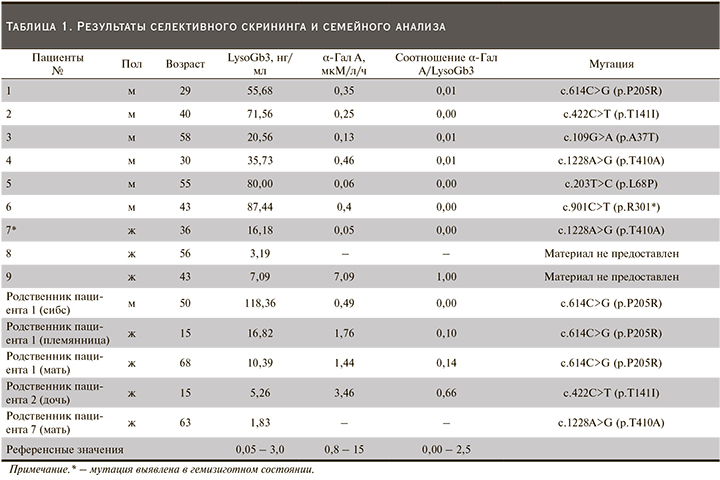
Активность фермента у всех обследованных пациентов мужского пола была значительно снижена. У всех женщин, выявленных при проведении селективного скрининга и семейного анализа, активность фермента была в пределах референсных значений, кроме пациентки 7.
Для подтверждающей диагностики применяли прямое секвенирование кодирующих экзонов гена и прилегающих к ним участков интронов. Выявлено 7 мутаций, большинство из которых описано ранее в литературе (у пациентов с классической формой БФ). Три мутации новые, обнаруженные в ходе данного исследования. Патогенность мутаций подтверждена на основании анализа in silico по программам MutationTaster, PolyPhen2, SIFT, PROVEAN, Human Splicing Finder.
У пациентки 7 36 лет, которая была родной сестрой пациента 4, при проведении ДНК-анализа обнаружена мутация с.1228A>G в гемизиготном состоянии. Анализ кариотипа пациентки показал наличие дополнительно к БФ синдрома Шерешевского–Тернера.
Обсуждение
Пациенты с патологией почек входят в группу риска БФ, поэтому проведение скрининга на это заболевание в диализных центрах и отделениях нефрологии распространены повсеместно. Результаты некоторых программ скрининга на БФ приведены в табл. 2. Распространенность БФ при проведении этих исследований составляет от 0,0 до 1,17. Более высокие цифры распространенности получены при обследовании сравнительно небольшого числа пациентов: возможно, врачами осуществлялся более тщательный клинический отбор в группу риска. В более репрезентативных выборках распространенность ниже и составляет менее 0,5%, вероятно, она отражает более точную распространенность БФ среди пациентов нефрологических отделений.
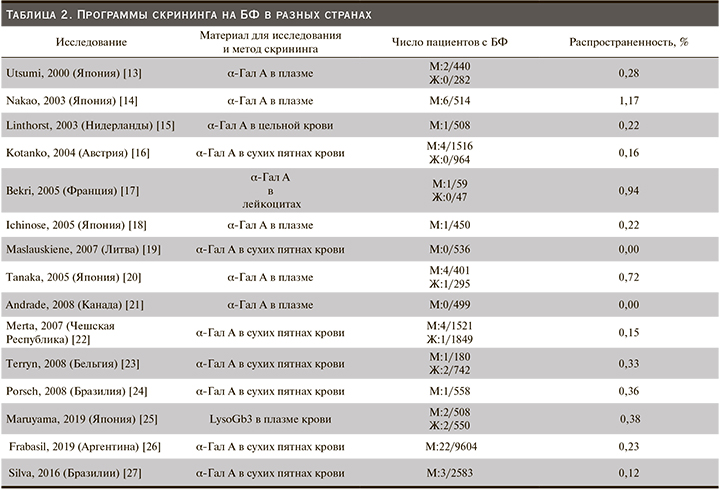
Распространенность БФ в отделениях гемодиализа в нашем исследовании составила 0,17%, что сопоставимо с исследованиями других стран, где выборка пациентов была достаточно велика.
Приблизительно до 2018 г. большинство программ селективного скрининга проводили с помощью определения активности α-Гал A. При БФ активность α-Гал A у мужчин снижена, у женщин может быть нормальной или около нижней границы нормы, что приводит к ложноотрицательным результатам. Сравнительно недавно показано, что концентрация LysoGb3 в плазме или высушенных пятнах крови повышается не только у мужчин, но и у женщин с БФ, что позволяет применять LysoGb3 в качестве биомаркера для диагностики и контроля лечения заболевания [25]. Данный биомаркер более чувствителен для диагностики женщин с БФ по сравнению с α-Гал A и всегда повышен у женщин с клиническими проявлениями заболевания, но его концентрация в крови женщин-носительниц ниже, чем у больных мужчин с БФ [10].
Интересен случай, выявленный в нашем исследовании: сочетание БФ и хромосомной патологии (синдром Шерешевского–Тернера), что объясняет высокий уровень LysoGb3 и довольно низкую активность фермента 0,05 мкМ/ч/л, что не характерно для женщин – носительниц БФ.
Поскольку БФ является Х-сцепленным наследственным заболеванием, немаловажно обследование родственников пробандов. Семейный скрининг позволяет выявлять пациентов на доклинической стадии заболевания [4, 18]. В нашем исследовании после проведения семейного анализа диагноз был дополнительно установлен пяти пациентам. К сожалению, не все пациенты готовы сообщать своим родным об установленном у них диагнозе наследственного заболевания, поэтому число прошедших тестирование не столь велико не только у нас, но и в других странах.
В целом скрининговое обследование находящихся на программном гемодиализе пациентов для обнаружения БФ позволяет выявлять новые случаи, особенно те, в которых поражение почек было ведущим проявлением данного заболевания.
Заключение
Таким образом, неспецифичные признаки поражения почек, наблюдаемые при БФ, зачастую усложняют верификацию диагноза и требуют от практикующих врачей специального подхода к оценке клинических симптомов и результатов лабораторной диагностики в каждом конкретном случае. Важно проведение семейного анализа, который позволяет выявлять пациентов с БФ на ранней стадии болезни и делает терапию более эффективной. Своевременное выявление поражения почек при проведении селективного скрининга в группах высокого риска с назначением в последующем специфической терапии позволяет стабилизировать функцию почек, снизить частоту развития поздних почечных осложнений.
Применение нового подхода к скринингу позволяет с большей эффективностью проводить исследование лиц как мужского, так и женского пола.
Полученные результаты позволяют рекомендовать определение концентрации LysoGb3 в пятнах высушенной крови уже на начальной стадии диагностического обследования пациентов с ХБП для исключения БФ.


