
Э озинофильный гранулематоз с полиангиитом (Черга–Страусс; ЭГПА) – это редко встречающийся АНЦА (антинейтрофильные цитоплазматические антитела)-ассоциированный системный васкулит, характеризующийся сочетанием бронхиальной астмы (БА) и эозинофилии (> 10 %), с поражением периферической нервной системы, кожи, сердца и других органов. В обзоре литературы рассматриваются варианты ЭГПА, методы диагностики и оценки активности и подходы к лечению.
В 1951 г. J. Churg и L. Strauss описали 13 пациентов с БА, эозинофилией и некротизирующим васкулитом сосудов преимущественно мелкого калибра, сочетавшимися с периваскулярной эозинофильной инфильтрацией и гранулематозом [1]. Большинство из этих пациентов находились под наблюдением с диагнозом узелкового полиартериита, однако с учетом наличия БА и эозинофильного гранулематоза, нехарактерных для этого диагноза, было предложено выделить отдельное заболевание – «аллергический ангиит с гранулематозом», который позже стали называть синдромом Черга–Страусс, а в настоящее время – эозинофильным гранулематозом с полиангиитом (ЭГПА) [1–3, 73]. В 1984 г. J. Lanham сформулировал критерии диагностики ЭГПА: БА, эозинофилия периферической крови > 1,5 × 109/л (или > 10 % от общего числа лейкоцитов) и признаки системного васкулита с поражением как минимум двух систем органов [2]. Однако наибольшее распространение получили классификационные критерии, разработанные в 1990 г. Американской коллегией ревматологов: БА, эозинофилия крови более 10 %, моноили полиневропатия, легочные инфильтраты, синуситы, экстраваскулярная эозинофильная инфильтрация, по данным биопсии [4]. Эти критерии основывались на результатах многофакторного анализа клинических, рентгенологических и гистологических данных 807 пациентов с системными васкулитами. Если у больного имеются по крайней мере 4 из 6 признаков, то диагностическая чувствительность составляет 85,0 %, специфичность – 99,7 % [5].
ЭГПА – один из наиболее редко встречающихся некротизирующих васкулитов. Распространенность и заболеваемость составляют соответственно 11–14 и 0,5–3,7 на 1 млн населения [6]. В литературе описаны случаи развития ЭГПА во всех возрастных группах, в т.ч. в детском и пожилом возрасте. Заболеваемость одинаковая для мужчин и женщин, хотя у последних отмечают развитие ЭГПА в более молодом возрасте, более длительный анамнез БА и меньшую частоту поражения почек [7].
Этиология и патогенез
В 1954 г. G. Godman и J. Churg предложили иммунную концепцию патогенеза ЭГПА, гранулематоза с полиангиитом (Вегенера) и узелкового полиартериита [8]. Первые данные, подтверждающие эту теорию, были получены в 1960-х гг., когда с помощью иммунофлуоресцентной микроскопии было установлено, что некоторые формы васкулита ассоциируются с отложением антител и комплемента в сосудистой стенке. Например, криоглобулинемический васкулит и пурпура Шенлейна–Геноха характеризуются образованием депозитов криоглобулинов и иммуноглобулина А (IgА) соответственно. Первоначально поражение сосудов при ЭГПА также связывали с отложением иммунных комплексов, хотя патогенез заболевания оставался неясным. Считалось, что триггером сосудистого повреждения при ЭГПА могут быть антигены микробов [9]. В пользу этой гипотезы свидетельствовали результаты ретроспективного исследования, в котором развитию ЭГПА нередко предшествовала вакцинация или терапия иммуномодуляторами. В 1982 г. у 8 больных гломерулонефритом, который в 5 случаях сочетался с поражением легких, были выявлены антитела к белкам цитоплазмы нейтрофилов (АНЦА) [10]. Вскоре после публикации была установлена связь этих антител c гранулематозом и полиангиитом (Вегенера) [11], а позднее – с ЭГПА [12]. Для обнаружения АНЦА применяют метод непрямой иммунофлуресценции – окрашивание фиксированных этанолом нейтрофилов здоровых добровольцев, которые инкубируются с сывороткой пациентов. Выделяют три варианта окрашивания гранулоцитов: цитоплазматический (цАНЦА), перинуклеарный (пАНЦА) и атипичный (любое положительное окрашивание, кроме первых двух). Для подтверждения наличия АНЦА проводят иммуноферментный анализ (ИФА). АНЦА связываются с компонентами цитоплазматических гранул нейтрофилов и моноцитов. Основной антигенной мишенью для цАНЦА является лизосомальная протеиназа (PR-3), для пАНЦА – миелопероксидаза (MPO). При ЭГПА частота выявления АНЦА, в основном к миелопероксидазе, составляет 30–40 % [7]. Взаимосвязь титра АНЦА и активности ЭГПА подробно не изучалась, поэтому сегодня их определение используют только с диагностической целью.
Открытие АНЦА стало важным этапом в развитии представлений о патогенезе ЭГПА. Доказательством их связи с развитием васкулита служит тот факт, что нейтрофилы в стенке воспаленных кровеносных сосудов активируют поверхностную экспрессию протеиназы 3 и миелопероксидазы [13, 14]. Связывание АНЦА с этими антигенами может приводить к дегрануляции нейтрофилов и повышению продукции ими супероксида, который вызывает повреждение сосудов. Взаимодействие АНЦА с эндотелием регулируется интерлейкином-8 (ИЛ-8), который является мощным индуктором хемотаксиса нейтрофилов и может усилить любой опосредованный нейтрофилами процесс сосудистого повреждения. Наконец АНЦА активируют выделение эндотелиальными клетками тканевого фактора, индуцирующего альтернативный путь свертывания крови и способствующего нарастанию сосудистого повреждения [15, 16]. Причины образования АНЦА и активации нейтрофилов при ЭГПА неизвестны.
В конце 1990-х гг. обсуждалась роль антагонистов рецепторов лейкотриенов (зафирлукаста) в развитии ЭГПА [17–20]. Высказывалось предположение, будто ЭГПА может быть проявлением реакции гиперчувствительности к препаратам этого класса. Однако при приеме антагонистов рецепторов лейкотриенов развитие ЭГПА обычно наблюдали после снижения дозы глюкокортикоидов (ГК), которые пациенты получали по поводу плохо контролируемой БА. Нельзя исключать, что в этих сравнительно немногочисленных случаях БА была первым проявлением ЭГПА, развитие которого провоцировали не антагонисты лейкотриеновых рецепторов, а снижение дозы ГК. В пользу этого предположения свидетельствует тот факт, что появление развернутой картины ЭГПА иногда наблюдали после отмены ГК у больных БА, не получавших антагонисты лейкотриеновых рецепторов [20]. Описаны также случаи развития ЭГПА при применении макролидных антибиотиков, карбамазепина, пропилтиоурацила, аллопуринола и хинина [21–25].
Говоря об ЭГПА, нельзя не отметить важную роль эозинофилов в развитии поражения сосудов. Многие авторы обращали внимание на корреляцию уровня эозинофилов и активности заболевания. С помощью моноклональных антител Р. Tai и соавт. продемонстрировали присутствие активированных эозинофилов и продуктов их дегрануляции как в стенке сосудов, так и в периваскулярных гранулемах [26]. Высвобождаемые эозинофилами медиаторы, такие как эозинофильный катионный белок и нейротоксин, оказывают кардиои нейротоксичное действия соответственно и вызывают развитие кардиомиопатии и полиневропатии. Процессы дифференцировки и активации эозинофилов, а также накопление их в тканях регулируются ИЛ-5 и такими специфическими хемокинами, как эотаксин-3 и CCL-17. ИЛ-5 (эозинофильный колониестимулирующий фактор) представляет собой полипептидный цитокин, продуцирующийся Т2-хелперами и тучными клетками. ИЛ-5 наряду с ИЛ-4 и ИЛ-13, которые также вырабатываются Т2-клетками, индуцируют дифференцировку В-клеток в плазматические клетки, что позволяет объяснить повышение уровня общего IgЕ, а также IgG4 при ЭГПА. Причиной повышения уровня ИЛ-5 считают дисрегуляцию Т-клеточного ответа с преобладанием Т2-звена под воздействием ИЛ-25, который в свою очередь вырабатывается эозинофилами («порочный круг») [27]. CCL17, скорее всего, выделяется дендритными клетками, уровень его коррелирует с числом эозинофилов [28].
В 2008 г. K. Polzer и соавт. [29] выявили значительное повышение уровня эотаксина-3 в сыворотке больных активным ЭГПА наряду с увеличением числа эозинофилов, уровня общего IgЕ и острофазовых показателей. В 2011 г. J. Zwerinа и соавт. [30] в более крупном исследовании также продемонстрировали связь повышения уровня эотаксина-3 с активностью ЭГПА. Эотаксин-3, продуцируемый эндотелиальными и воспалительными клетками в ответ на повреждение, считают чувствительным и специфическим (87,5 и 98,6 % соответственно при предельном уровне 80 пг/мл) маркером активности ЭГПА.
В настоящее время обсуждается генетическая предрасположенность к ЭГПА, ассоциированная с HLADRB1*04 и HLADRB*07 [31, 72]. Для АНЦА-негативного варианта ЭГПА установлена связь с гаплотипом ИЛ-10, 2 промотера гена ИЛ-10, что проявляется повышением уровня ИЛ-10 [32].
Клиническая картина ЭГПА
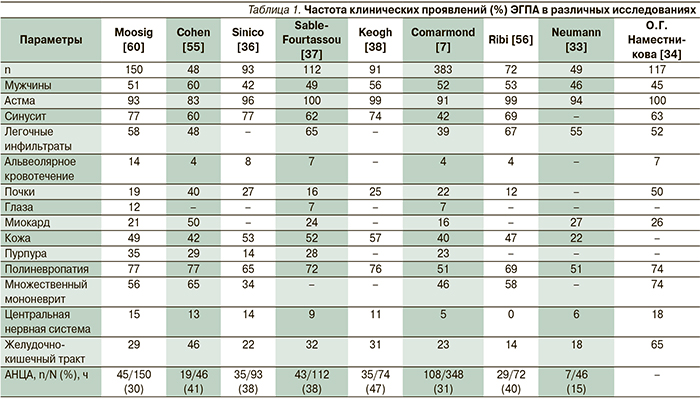
Заболевание обычно начинается с аллергического риносинусита с полипозом и БА, позднее появляются эозинофилия и эозинофильная инфильтрация тканей с последующим развитием васкулита (в среднем через 3–9 лет после начала болезни) (табл. 1). Однако иногда БА развивается после васкулита [33]. БА, обычно начинающаяся в зрелом возрасте и быстро прогрессирующая, наблюдается у 95–100 % больных ЭГПА, полипоз носа и синусит – приблизительно у 50 % [34]. Применение ГК может маскировать симптомы системного васкулита [19]. ЭГПА следует подозревать, если БА сопровождается высокой эозинофилией. Более чем у половины пациентов встречаются неспецифические симптомы, такие как слабость, снижение массы тела, миалгии, артралгии или артрит [3]. У 37–77 % больных поражаются легкие [35]. У 75–81 % больных ЭГПА развивается множественный мононеврит [36–38]. Чаще всего поражаются общий малоберцовый и внутренний подколенный нервы, что клинически проявляется провисанием стопы, нарушением чувствительности, болью [39, 40].
Для подтверждения диагноза может быть выполнена биопсия нерва, хотя признаки некротизирующего васкулита и периневральную инфильтрацию эозинофилами и воспалительными клетками выявляют только в половине случаев, а чаще всего определяется дегенерация аксонов. Поражение центральной нервной системы встречается реже и ассоциируется с более высокой летальностью [14]. Описаны поражение 2-й, 3, 7 и 8-й пар черепномозговых нервов и ишемия зрительного нерва [3]. Иммуносупрессивная терапия может приводить к полному обратному развитию неврологической симптоматики. У 25 % больных отмечается поражение кожи в виде пурпуры, сетчатого ливедо, подкожных узелков, уртикарной сыпи и кожных язв [41].
Выделяют два основных варианта поражения сердца при ЭГПА – эозинофильный миокардит, который может приводить к развитию кардиомиопатии рестриктивного или дилатационного типа, и коронариит, проявляющийся ишемией миокарда. Важную роль в патогенезе поражения миокарда играют токсические медиаторы эозинофилов. При ЭГПА описаны также перикардит и поражение клапанов [42].
Фокальный и сегментарный некротизирующий гломерулонефрит – это наиболее распространенный вариант поражения почек при ЭГПА, встречающийся у 20–47 % пациентов [2, 6]. Могут наблюдаться эозинофильная инфильтрация канальцев, гранулематоз и васкулит. Почечная недостаточность развивается редко [1].
Поражение желудочно-кишечного тракта – ЖКТ (эозинофильный гастроэнтерит или васкулит мезентериальных сосудов) отмечается у 23 % больных [7]. Характерные симптомы – боль в животе, реже развиваются кишечная непроходимость, тошнота, рвота, диарея и кровотечение.
Необходимо отметить, что клинические особенности ЭГПА во многом определяются АНЦА-статусом [36, 37]. C. Comarmond и соавт. [7], которые наблюдали 383 пациента с ЭГПА в течение в среднем более 5 лет, продемонстрировали более частое поражение кожи, периферической нервной системы, почек и ЛОР-органов у АНЦА-положительных больных, в то время как при АНЦА-негативном варианте заболевания чаще встречалось поражение сердца. Кроме того, авторы отметили более высокую частоту обострений (35,2 против 22,5 % соответственно, р = 0,01), но более низкую смертность (5,6 против 12,5 %; р < 0,05) у АНЦА-положительных больных. Независимыми предикторами смерти были кардиомиопатия и пожилой возраст, а предиктором обострений – невысокое число эозинофилов в дебюте заболевания.
Диагноз и оценка активности
ЭГПА следует подозревать у пациентов среднего возраста с длительно текущей БА, аллергическим ринитом и эозинофилией при развитии у них признаков системного заболевания, включающих множественный мононеврит, легочные инфильтраты, кардиомиопатию, кожную пурпуру [43]. Большое диагностическое значение имеет наличие АНЦА, чаще к миелопероксидазе. На рентгенограмме грудной клетки обычно определяются инфильтративные изменения, часто преходящие [44]. S. Worthy и соавт. [45] при компьютерной томографии выявили инфильтрацию паренхимы легких, преимущественно в периферических отделах, у 15 (87 %) из 17 больных ЭГПА. Реже встречались плевральный и перикардиальный выпот, очаговые уплотнения с полостями, утолщение стенок бронхов и междольковых перегородок. Для диагностики поражения сердца применяют эхокардиографию [46]. Информативным методом диагностики кардиомиопатии считают магнитно-резонансную томографию (МРТ), которая позволяет оценивать структурные и функциональные особенности, перфузию и рубцовые изменения миокарда. Для дифференциальной диагностики рубцовых и воспалительных изменений миокарда предлагают использовать позитронноэмиссионную томографию или МРТ с контрастированием гадолинием [47].
К лабораторным признакам активности ЭГПА относят увеличение числа эозинофилов, а также С-реактивного белка и СОЭ. Как указано выше, обсуждается роль более специфичных маркеров активности, таких как уровень эотаксина-3. Для клинической оценки активности ЭГПА, как и других системных васкулитов, используют индекс BVAS (Birmingham Vasculitis Activity Score), в то время как тяжесть необратимых изменений определяют с помощью индекса VDI (Vasculitis Damage Index) [48].
Дифференциальный диагноз
При обследовании пациентов с предполагаемым диагнозом ЭГПА необходимо исключить другие причины гиперэозинофилии и подтвердить наличие васкулита. В 1936 г. W. Lоeffler описал случаи «легочной эозинофилии» у пациентов с возвратным лимфангитом, лимфатической обструкцией и летучими инфильтратами в легких в ответ на инфекцию, вызванную Wuchereria bancrofti [49]. Причинами эозинофильной пневмонии помимо ЭГПА могут быть аллергический бронхолегочный аспергиллез, паразитарные инвазии (токсокароз и стронгилоидоз). В пользу ЭГПА свидетельствуют системные проявления, характерные для васкулита, в частности поражение нервной системы, почек и т.д. Важную роль в диагностике ЭГПА отводят гистологическому исследованию биоптата, например, кожи или почек с целью обнаружения признаков васкулита [50]. Определенные трудности возникают при проведении дифференциального диагноза ЭГПА с гиперэозинофильным синдромом, который представляет собой неоднородную группу состояний (миелопролиферативный и лимфопролиферативный варианты, семейный, идиопатический, ассоциированный с аутоиммунными заболеваниями, перекрестный), характеризующихся персистирующей эозинофилией более 1,5 × 109/л в течение не менее 6 месяцев. Миелои лимфопролиферативные варианты гиперэозинофильного синдрома развиваются в результате клональной пролиферации эозинофилов и Т-лимфоцитов соответственно, продуцирующих эозинофилопоэтины (ИЛ-5). Гиперэозинофильный синдром чаще встречается среди молодых мужчин и характеризуется интерстициальной инфильтрацией легких, тромбоэмболиями ветвей легочной артерии, тяжелым поражением сердца с застойной сердечной недостаточностью, резистентностью к терапии ГК [51, 52]. Уровень эотаксина-3 более чем у половины больных нормальный в отличие от больных ЭГПА [29]. Ключевое значение для дифференциальной диагностики имеют результаты исследования биоптата костного мозга [52]. Получены предварительные результаты, согласно которым наличие гена HLA-DRB4 у пациентов с БА связано с высоким риском развития симптомов продромального периода ЭГПА, что позволяет обсуждать возможности прогнозирования его развития до генерализации васкулита [77], однако эти данные требуют дальнейшего подтверждения.
Прогноз и лечение
Прогноз для жизни при ЭГПА в целом относительно благоприятный. Иммуносупрессивная терапия обычно позволяет добиваться ремиссии заболевания, хотя у части больных развиваются рецидивы, необратимое поражение органов и тканей, в частности сердца или периферической нервной системы, и смерть. Во французском когортном исследовании (n = 348) в течение в среднем 5 лет частота рецидивов васкулита составила 25,3 %, в т.ч. 35,2 и 22,5 % у больных АНЦАпозитивным и АНЦА-негативным ЭГПА соответственно (р = 0,01). Следует отметить, что еще у 18,8 % пациентов во время наблюдения отмечались обострения БА, синусита и/или нарастание эозинофилии, обосновавшие модификацию терапии. В течение того же срока умерли 11,7 % больных, в т.ч. 5,6 % больных АНЦА-позитивным вариантом заболевания и 12,5 % больных АНЦА-негативным. У трети больных причиной смерти были сердечнососудистые осложнения (инфаркт миокарда, сердечная недостаточность или аритмии), которые обычно развивались в отдаленном сроке после начала ЭГПА [7]. Реже больные умирали от инфекционных заболеваний, злокачественных опухолей, активного васкулита, легочных осложнений и других причин.
Общепринятых рекомендаций по лечению ЭГПА нет. Схему иммуносупрессивной терапии ЭГПА следует подбирать индивидуально с учетом активности васкулита и тяжести висцеральных проявлений. Желательно, чтобы пациент находился под наблюдением ревматолога, имеющего опыт лечения больных системными васкулитами. Цель терапии – добиться полной ремиссии заболевания (отсутствие активности системного васкулита и доза преднизолона ≤ 7,5 мг/сут; если не удается снизить дозу преднизолона до указанного значения, результат терапии расценивают как частичную ремиссию). Следует учитывать, что сохранение «остаточных» проявлений системного васкулита, например полиневропатии или кардиомиопатии, не исключает ремиссию заболевания. Отсутствие динамики указанных изменений и новых проявлений васкулита в течение ≥ 3 месяцев указывает на отсутствие активности заболевания (в этом случае индекс BVAS равен 0 даже при наличии достаточно тяжелых висцеральных проявлений). У большинства больных ЭГПА, ответивших на иммуносупрессивную терапию, сохраняются симптомы БА.
Для индукции ремиссии всем больным следует назначать ГК в дозе 0,5–1,0 мг/кг. При высокой активности заболевания лечение целесообразно начинать с пульс-терапии ГК (метилпреднизолон в дозе 1 г внутривенно в течение 3 дней). Эксперты Французской группы по изучению васкулита (French Vasculitis Study Group) [53, 54] рекомендуют добавлять иммунодепрессанты (циклофосфамид или азатиоприн) к ГК только при наличии неблагоприятных прогностических факторов или отсутствии ответа на монотерапию. В нескольких достаточно крупных исследованиях, которые проводились во Франции, целесообразность включения циклофосфамида в схему индукционной терапии оценивали с учетом индекса FFS (Five Factors Score), который был предложен в 1996 г. на основании когортного исследования больных ЭГПА и узелковым полиартериитом и включает поражение сердца, центральной нервной системы, ЖКТ, а также сывороточный уровень креатинина > 140 мкмоль/л и суточную протеинурию > 1 г/сут [55]. C. Ribi и соавт. [56] проводили монотерапию ГК и 72 больным ЭГПА, у которых отсутствовали неблагоприятные прогностические факторы (FFS = 0). Лечение ГК позволило достичь ремиссии 93 % больных, однако у трети из них при снижении дозы развился рецидив, в основном в течение первого года после начала терапии, что потребовало назначения иммунодепрессантов. Кроме того, у трети больных наблюдались побочные эффекты глюкокортикоидной терапии. Авторы сопоставили также эффективность азатиоприна и циклофосфамида для 19 больных, у которых отсутствовал ответ на глюкокортикоидную терапию или развился рецидив. Ремиссия была достигнута 50 и 78 % больных соответственно, однако разница не достигла статистической значимости. Хотя исследование подтвердило прогностическое значение индекса FFS (выживаемость через 1 и 5 лет составила 100 и 97 % соответственно), однако высокая частота рецидивов определяет необходимость изучения других предикторов ответа на монотерапию ГК.
В отношении 48 больных ЭГПА и по крайней мере одним неблагоприятным прогностическим фактором (FFS ≥ 1) Е. Cohen и соавт. [55] сравнивали эффективность 6 и 12 сеансов пульстерапии циклофосфамидом в дозе 0,6 г/м2 (каждые 2 недели в течение первого месяца, а затем каждые 4 недели). Ремиссия была достигнута в 87,5 % случаев. Частота ее была сопоставимой в двух группах (91,3 и 84,0 % соответственно). В течение 8-летнего наблюдения рецидивы имели место у 64,6 % больных, однако большинство из них были легкими. Общая частота рецидивов и «больших» рецидивов (требовавших возобновления терапии иммунодепрессантами) была сопоставимой в группах больных, получивших 6 или 12 сеансов пульс-терапии циклофосфамидом, однако легкие рецидивы (требовавшие только временного увеличения дозы ГК) чаще развивались после завершения более короткой терапии циклофосфамидом. С учетом преимущества длительной терапии этим препаратом набор больных в исследование был прекращен досрочно. Результаты этого исследования показали целесообразность длительной комбинированной терапии ГК и иммунодепрессантами больных с неблагоприятными прогностическими факторами, хотя для поддерживающей терапии вместо циклофосфамида лучше использовать менее токсичные азатиоприн или, возможно, метотрексат, эффективность которых установлена при других АНЦА-ассоциированных васкулитах, в частности гранулематозе с полиангиитом (Вегенера) [57].
Следует отметить, что практическое значение индекса FFS для выбора тактики иммуносупрессивной терапии вызывает сомнение [74]. Во-первых, этот индекс не специфичен для ЭГПА, т.к. был разработан при анализе течения различных васкулитов. При этом эффективность монотерапии ГК для больных со значением FFS, равным 0, в исследовании C. Ribi и соавт. [56] была ограниченной. Во-вторых, в 2011 г. FFS был пересмотрен на основании результатов когортного исследования 1108 больных ЭГПА и другими васкулитами [58]. Пересмотренный индекс предлагают рассчитывать с учетом следующих 5 факторов: возраст старше 65 лет, поражение сердца или ЖКТ, сывороточный уровень креатинина ≥ 150 мкмоль/л и отсутствие поражения ЛОР-органов. Во французском когортном исследовании у 348 первичных больных ЭГПА значение «оригинального» FFS было равным 0 у 76,0 % больных, а пересмотренного – у 25,6 %. Таким образом, доля пациентов, «нуждающихся» в назначении иммунодепрессантов с учетом значения FFS ≥ 1, при использовании двух вариантов индекса отличалась в 3 раза. В том же исследовании комбинированная индукционная терапия ГК и иммунодепрессантами проводилась более чем половине больных, а на протяжении всего 5-летнего наблюдения применение по крайней мере одного иммунодепрессанта потребовалось в 71 % случаев (чаще всего применяли циклофосфамид и азатиоприн). Кроме того, при расчете FFS не учитываются некоторые проявления ЭГПА, например поражение периферической нервной системы, которые, возможно, не позволяют оценивать вероятность летального исхода, но могут определять рефрактерность к терапии ГК. Например, в исследовании C. Ribi и соавт. [56] периферической невропатией страдали 37 % больных, не ответивших на монотерапию ГК, а поражение кожи – 27 %. Необходимо помнить, что кардиомиопатия, учитывающаяся при расчете FFS, для больных ЭГПА может быть следствием не только системного васкулита, но и атеросклероза [59].
Более обоснованной представляется стратегия лечения больных ЭГПА, предложенная F. Moosig и соавт. [60], которые считали показанием к назначению циклофосфамида не только FFS ≥ 1, но и наличие висцеральных проявлений, которые создавали угрозу для жизни или могли привести к нарушению функции соответствующего органа. К последним отнесли альвеолярное кровотечение, тяжелый эозинофильный альвеолит, гломерулонефрит, миокардит или коронариит, поражение периферической или центральной нервной системы, ЖКТ (диарея с примесью крови, которую нельзя было объяснить другими причинами и/или гистологические данные). В таких случаях больным назначали не только преднизолон (1 мг/кг), но и циклофосфамид (2 мг/кг внутрь в течение 3 месяцев или в виде пульстерапии по 15 мг/кг, n = 6). Дозу преднизолона начинали снижать через 3–8 дней после начала лечения (цель – ≤15 мг/сут через 2–3 месяца и ≤ 7,5 мг в последующем). В случае достижения ремиссии больных переводили на поддерживающую терапию метотрексатом (10–30 мг/сут), лефлуномидом (10–30 мг/сут) или азатиоприном (2 мг/кг). В отсутствие угрожающих висцеральных проявлений проведена монотерапия преднизолоном в дозе от 15 до 75 мг/сут. Если в течение 3 месяцев дозу препарата не удавалось снизить до 7,5 мг/сут, присоединяли указанные выше иммунодепрессанты. Поддерживающую терапию в ряде случаев прекращали, если ремиссия сохранялась в течение по крайней мере года. Применение циклофосфамида потребовалось 71 % из 150 больных ЭГПА, включенных в данное исследование. В целом ремиссия была достигнута примерно у 90 % больных, остававшихся под наблюдением, в т.ч. полная – у 67 %. Медиана дозы преднизолона в конце наблюдения составила всего 5 мг/сут. В течение 4-летнего наблюдения большие рецидивы зарегистрированы у 14 % больных, а расчетная 5и 10-летняя выживаемость достигла 97 и 89 %. Таким образом, в этом исследовании более агрессивная иммуносупрессивная терапия, предполагавшая более частое применение циклофосфамида на начальном этапе лечения и других иммунодепрессантов на этапе поддерживающей терапии, позволила достичь ремиссии подавляющему большинству больных ЭГПА и обеспечила приемлемую частоту больших рецидивов, высокую 5и 10-летнюю выживаемость.
При неэффективности стандартной индукционной терапии за последние годы к больным системными васкулитами обычно применяют генноинженерные биологические препараты [61, 62]. В небольших сериях наблюдений больным рефрактерным ЭГПА показана эффективность ритуксимаба, ингибиторов фактора некроза опухоли α, а также внутривенного иммуноглобулина и интерферона α [63–66]. В то же время плазмаферез не улучшал результаты иммуносупрессивной терапии больных ЭГПА [67]. Перспективным препаратом считают меполизумаб, разработанный для лечения БА [68]. Меполизумаб представляет собой гуманизированные моноклональные антитела (IgG1) к ИЛ-5, которые подавляют взаимодействие этого цитокина с α-цепью соответствующих рецепторов. В двух небольших сериях случаев (7–10 пациентов) применение меполизумаба оказывало стероидосберегающее действие или позволяло достигать ремиссии большинству больных стероидозависимым или рефрактерным ЭГПА [69, 70] Однако эффективность меполизумаба, как и других генно-инженерных биологических препаратов, необходимо подтвердить в более крупных исследованиях [75, 76].
Заключение
ЭГПА – это АНЦА-ассоциированный системный васкулит, который обычно развивается у больных БА в среднем возрасте, характеризуется эозинофилией и чаще всего поражает нервную систему и кожу. Хотя 5и 10-летняя выживаемость при ЭГПА высокая, заболевание нередко рецидивирует и требует длительной (часто пожизненной) иммуносупрессивной терапии. Всем пациентам с ЭГПА следует назначать ГК, которые сочетают с циклофосфамидом при наличии неблагоприятных прогностических факторов (FFS ≥ 1) или тяжелых висцеральных проявлений. Для поддерживающей терапии иммунодепрессанты (прежде всего азатиоприн) обычно применяют при тяжелом или рецидивирующем течении системного васкулита, а также при стероидозависимой форме заболевания (т.е. при невозможности снижения дозы преднизолона ≤ 7,5 мг/сут).


