
Псевдогипопаратиреоз (ПГП) и наследственная остеодистрофия Олбрайта (НОО) – очень редкие и гетерогенные заболевания с доказанным генетическим компонентом, распространенность которых составляет 7,9 на 1 млн человек [1]. Исторически ПГП – первый синдром, вызванный резистентностью к гормонам, он был описан Олбрайтом и соавт. в 1942 г. [2]. В настоящее время термин охватывает высокогетерогенную группу редких метаболических нарушений, каждое из которых характеризуется резистентностью органа-мишени к ПТГ. Олбрайт описал пациентов с нормальной функцией почек, у которых гипокальцемия и гиперфосфатемия сочетались с более низкой экскрецией кальция и фосфора с мочей в ответ на введение ПТГ по сравнению с пациентами при первичном гипопаратиреозе. Причиной таких результатов Олбрайт посчитал наличие резистентности к ПТГ. В последующих исследованиях пациентов с ПГП была описана гиперплазия паращитовидных желез и повышение уровня иммунореактивного ПТГ, что косвенно подтверждает резистентность, лежащую в основе заболевания [3, 4]. После идентификации рецептора к ПТГ было выявлено нарушение проведения сигнала от рецептора в клетках-мишенях [5] при ПГП I типа, тем самым был подтвержден факт наличия резистентности. Молекулярный дефект, вызывающий ПГП II типа, в настоящий момент не выявлен, поэтому это диагноз-исключение в отсутствие данных за I тип.
Патогенез резистентности к ПТГ
ПТГ – основной регулятор сывороточной концентрации кальция, органами-мишенями ПТГ являются почки и кости, где он действует через G-сопряженный рецептор рПТГ-1 [6, 7]. В норме ПТГ стимулирует транскрипцию гена 25-гидроксивитамин Д1-гидроксилазы в почечных проксимальных канальцах, что ведет к образованию активного 1,25-дигидроксивитамина Д3, который в свою очередь усиливает абсорбцию кальция и фосфатов из кишки. В проксимальных канальцах почки ПТГ препятствует реабсорбции фосфатов, а в дистальном нефроне усиливает реабсорбцию кальция [8].
Дефект ответа почек на ПТГ – это главная характеристика всех видов ПГП. Резистентность происходит только в проксимальных канальцах нефронов, что приводит к задержке фосфатов. Действие ПТГ на дистальные канальцы неизменно, поэтому антикальциурическое действие ПТГ остается нетронутым у всех пациентов с ПГП, что объясняет нормокальциурию, гиперфосфатемию, сохранную почечную функцию в течение всей жизни и отсутствие конкрементов почек [9]. Нарушение чувствительности к ПТГ обусловлено генетическим дефектом в локусе GNAS.
Локус GNAS находится в хромосоме 20q13, он является одним из самых сложно устроенных среди всех эукариотических клеток и содержит информацию об алгоритмах импринтинга, альтернативного сплайсинга, транскрипции антисмысловых РНК с тканевой и возрастспецифичной экспрессией. Таким образом, в соответствии с унаследованным генотипом из локуса GNAS транскрибируется несколько генов: субъединица гетеротримерного стимулирующего G-белка (Gs), экстрабольшой вариант Gs (XLs) и альтернативный продукт гена – нейроэндокринный протеин 55 (НЭП55). Транскрипт G-белка экспрессируется билатерально, за исключением проксимальных почечных канальцев, щитовидной железы, гонад и гипоталамуса; их экспрессия определяется материнской хромосомой. Пациенты с ПГП стадают дефектом этого локуса (мутация или нарушение метилирования) [10], который приводит к синтезу дефектного G-белка или нарушает импринтинг всего локуса. Кроме самого дефекта имеет значение место и время начала экспрессии GNAS, которое крайне вариабельно. ПГП развивается, только если локус GNAS передается от матери, тип наследования при этом аутосомно-доминантный [11–13]. В силу того что материнский локус GNAS отвечает за экспрессию G-белка в щитовидной железе, гонадах и гипоталамусе пациентов с ПГП Ia, наблюдается резистентность к другим гормонам, чьи рецепторы сопряжены с G-белком, таким как ТТГ, ЛГ и ФСГ; нарушения варьируются по выраженности и продолжительности [14–17]. Это объясняет высокое разнообразие клинической картины и времени манифестации ПГП.
Клиническая картина НОО и ПГП
НОО – это клинический синдром, объединяющий разнообразные клинические проявления, такие как брахидактилия, лунообразное лицо, низкий рост, центральное ожирение, подкожная оссификация и разная степень умственной отсталости [18, 19]. К самым характерным клиническим признакам НОО относится укорочение III, IV, V пястных костей и I дистальной фаланги в сочетании с гетеротопической оссификацией. Умственная отсталость была изначально описана как обязательный компонент НОО. В отличие от других признаков при последующих исследованиях не удалось достоверно доказать ее специфичность для синдрома, тем не менее ее распространенность среди пациентов – взрослых и детей – составляет 27 и 64 % соответственно [19, 20].
Причиной развития НОО служит ПГП, различают несколько типов: Iа, Ib, Ic и II. Каждый тип имеет свои особенности, подробнее всего описан тип Iа. Резистентность к ПТГ при ПГП вызывает гипокальцемию, гиперфосфатемию и повышение уровня ПТГ. Резистентность к ПТГ развивается чаще в первые годы жизни. Сначала происходит повышение ПТГ и фосфатов, затем снижение уровня кальция. В то же время при обследовании пациентов минеральная плотность костей сохраняется нормальной и даже повышенной в группах детей и взрослых [21]. Более того, пациенты с ПГП также имеют нормальный или сниженный риск переломов по сравнению с популяцией. Эти данные косвенно свидетельствуют о резистентности костей к ПТГ.
Для ПГП Ib характерно более мягкое течение по сравнению с Iа, обусловленное изолированной почечной резистентностью к ПТГ без проявлений НОО и резистентности к другим гормонам [22–25]. ПГП Iс отличается от Iа менее выраженной резистентностью к ПТГ из-за неполного (50 %) снижения активности G-белка, сопряженного с рецепторами. Несмотря на наличие определенных особенностей каждого типа ПГП, внутри типов отмечается широкий диапазон вариантов клинических проявлений, поэтому сложно дифференцировать, какой именно тип ПГП присущ конкретному пациенту; в настоящее время рассматривается вопрос о совершенствовании классификации исходя из данных, полученных за последнее время.
Диагностика ПГП
Клиническая картина ПГП разнообразна, и в то же время сочетание симптомов довольно специфично, поэтому может служить одним из критериев диагностики. При наличии клинической картины ПГП следует проводить анализ крови на общий и ионизированный кальций, неорганический фосфор и ПТГ. При ПГП кальций общий и ионизированный окажутся сниженными, неорганический фосфор и ПТГ – повышенными. Эти результаты в сочетании с клинической картиной ПГП подтверждают диагноз. В случае мягкого течения или случайной лабораторной находки следует исключать дефицит витамина Д и почечную недостаточность. При наличии клинической картины НОО и отсутствии выраженных изменений при анализе крови есть вероятность наличия у пациента ПГП Iс или ПГП II, для дифференциальной диагностики в данном случае необходимо использовать генетический тест на дефекты локуса GNAS.
Лечение ПГП
Методом лечения для всех видов ПГП служит терапия витамином Д и кальцием (при гипокальцемии), цель терапии – нормализация кальция крови и ПТГ в кратчайшие сроки. Также стоит помнить, что у этих пациентов крайне низок риск повышения экскреции кальция с мочей на фоне терапии кальцитриолом или альфакацидолом, поэтому их доза может быть гораздо выше обычной без развития токсичности [8].
Клинический случай
Пациент – мужчина 33 лет европеоидной расы предъявляет жалобы на эпизоды потери сознания с 2000 г.
Потеря сознания происходит внезапно без каких-либо предвестников, при восстановлении сознания пациент помнит все, что было до момента приступа. Частота возникновения приступов – около 1 раза в год. Со слов свидетелей после потери сознания у пациента развивались выраженные судороги. В 2002 г. в связи с переутомлением проходил стационарное лечение, при обследовании диагностирован синдром Фара. Последний эпизод потери сознания произошел в 2008 г. на работе в ночную смену. Был госпитализирован в неврологическое отделение городской клинической больницы. При осмотре отмечено наличие стигм дисэмбриогенеза: скошенный затылок, низкопосаженные уши и близкопосаженные глаза. В неврологическом статусе отмечены следующие отклонения: симметричный положительный симптом Бабинского, тонус мышц изменен по пирамидно-экстрапирамидному типу. Клинический анализ крови и мочи, биохимический анализ крови (общий белок, мочевина, холестерин, билирубин, АЛТ, АСТ, глюкоза) без отклонений от референсных значений. Выписан с «диагнозом синдром Фара».
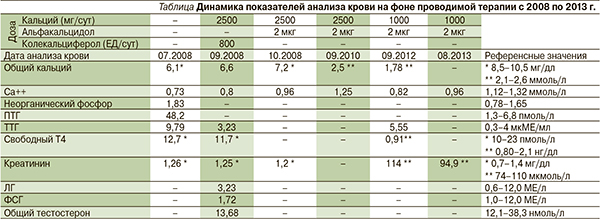
Для обследования и лечения 07.08 пациент был госпитализирован в специализированный неврологический стационар. В неврологическом статусе были выявлены следующие отклонения: непостоянный мелкоразмашистый нистагм в крайних отведениях, мандибулярный рефлекс слегка оживлен, сухожильные рефлексы симметричны, торпидные. При КТ и МРТ головного мозга выявлены асимметрия боковых желудочков, массивные обызвествления в подкорковых лобных и теменных ганглиях. Данных за нарушение кровообращения по результатам ультразвуковой допплерографии магистральных артерий головы получено не было. По результатам ЭЭГ отмечена выраженная дисфункция срединных неспецифических структур головного мозга, при гипервентиляции – с легкими признаками снижения по рога судорожной готовности. УЗИ щитовидной железы выявило умеренные диффузные изменения ткани щитовидной железы, паращитовидные железы не визуализировались. При анализе крови отмечено снижение уровня общего и ионизированного кальция, повышение уровня неорганического фосфора, ПТГ и ТТГ (см. таблицу).
Пациент был проконсультирован эндокринологом: диагностирован псевдогипопаратиреоз, рекомендован прием препаратов кальция (2500 мг/сут) в сочетании с витамином Д3 (800 ЕД/сут) под контролем кальция крови. По результатам консультации генетика: фенотип соответствует псевдогипопаратиреозу, рекомендована госпитализация в эндокринологический стационар для дообследования и подбора терапии.
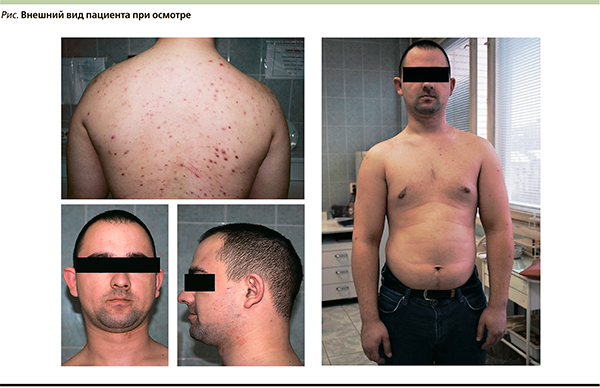
Пациент был госпитализирован в эндокринологический стационар 09.08. При осмотре: рост 182, вес 97 кг, индекс массы тела = 29,28 кг/м2. Обращают на себя внимание лунообразное лицо, кожные покровы с пустулезными высыпаниями на спине, бледно-розовые умеренной влажности, подкожно-жировая клетчатка развита избыточно, распределена равномерно (см. рисунок). Симптомы Хвостека и Труссо отрицательные. Щитовидная железа при пальпации не увеличена, узлы не пальпируются.
При обследовании было подтверждено наличие ПГП на основании осмотра (стигмы дизэмбриогенеза, подкожная оссификация), судорожного синдрома в анамнезе, снижения кальция крови и повышения ПТГ. При анализе крови от 22.09.08 отмечена положительная динамика (см. таблицу). Было рекомендовано продолжить терапию препаратами кальция (2,5 г/сут) и заменить витамин Д3 активным метаболитом: альфакальцидолом 2 мкг/сут. На фоне проводимой терапии уровень ионизированного кальция вырос до 0,96 ммоль/л (см. таблицу). Согласно имевшимся клиническим и лабораторным данным, был диагностирован ПГП Iа. По результатам генетического анализа локуса GNAS подтвержден ПГП Iа. Пациенту рекомендовано наблюдение эндокринолога и разъяснена необходимость регулярного контроля биохимических показателей крови.
В дальнейшем по рекомендации эндокринолога пациент контролировал уровень кальция и креатинина крови, а также ТТГ. После достижения нормального значения ионизированного кальция крови (см. таблицу) в 2010 г. доза препаратов кальция была снижена до 2000 мг/день, доза альфакальцидола не изменена, при следующем плановом анализе крови рекомендован контроль ПТГ. Однако до 2012 г. пациент не контролировал уровень кальция крови, ТТГ и ПТГ.
В начале 2012-го пациент самостоятельно снизил дозу препаратов кальция до 1000 мг/сут, в результате уровень ионизированного кальция снизился (см. таблицу, анализ крови от 09.12). При обращении к врачу в 2013 г. после анализа крови от 08.13 рекомендовано возобновить прием 2000 мг кальция/сут и продолжить прием альфакальцидола в прежней дозе. Обращает на себя внимание также наличие у пациента субклинического гипотиреоза, в связи с чем рекомендован контроль ТТГ и свободного Т4. Пациенту была повторно разъяснена необходимость регулярного контроля биохимических показателей крови.
Эпизодов потери сознания и судорожного синдрома после диагностики и начала лечения не отмечено. В настоящее время пациент трудоустроен и в его семье планируется прибавление.


