
Распространенность и причины анемии у пациентов с хронической сердечной недостаточностью
Несмотря на значительные успехи, достигнутые в лечении больных, хроническая сердечная недостаточность (ХСН) ассоциируется с высокими показателями заболеваемости и смертности. Кроме того, в настоящее время ХСН уже не являются исключительно “кардиологической” проблемой и приобретает ряд междисциплинарных аспектов для специалистов различных областей клинической медицины. Все большее внимание исследователей привлекает сочетание ХСН и анемии, которая зачастую лечится неадекватно или вообще не корригируется, а порой даже не выносится в диагноз. Между тем анемия вносит потенциальный вклад в развитие и прогрессирование ХСН [1].
Распространенность анемии среди пациентов c ХСН, по разным данным, составляет от 10 до 50 %. Такой большой разброс показателей можно объяснить отсутствием единого подхода к диагностике анемий, различиями в возрастном и половом составе пациентов [2], наличием сопутствующей патологии, в частности хронической почечной недостаточности, артериальной гипертензии [3] и степенью тяжести ХСН [4]
Причины и патогенетические механизмы анемии при ХСН неоднозначны. По данным J. Ezekowitz, у 58 % пациентов встречается анемия хронических заболеваний (АХЗ), у 21 % – железодефицитная анемия (ЖДА) [3]. Согласно сведениям J.N. Nanas, ЖДА диагностирована в 73 % случаев, АХЗ – в 18,9 %, у 5,7 % пациентов выявлена гемодилюция, а у 2,4 % – анемия трактовалась как следствие приема лекарств [5].
Дефицит витамина В12 и фолиевой кислоты встречается у пациентов с ХСН довольно редко [6]. Дефицит железа среди больных ХСН диагностировался, по разным данным, в 5–21 % случаев [3, 5, 7, 8]. ЖДА у больных ХСН может развиваться в результате синдрома мальабсорбции и скрытых желудочно-кишечных кровотечений, вызванных приемом ацетилсалициловой кислоты. В исследовании R. de Silva и соавт. показано, что в 43 % случаев встречается снижение концентрации либо сывороточного железа, либо ферритина, однако микроцитарная анемия обнаруживается лишь в 6 % случаев [9]. В то же время, согласно данным J.N. Nanas и соавт., снижение запаса железа в костном мозге обнаруживается у 73 % пациентов. При этом уровень сывороточного железа, ферритина и эритропоэтина (ЭПО) в сыворотке оставался в пределах нормы, а средний объем эритроцита соответствовал нижней границе нормы, что не укладывалось в микроцитарный характер анемии [5]. Эти данные свидетельствуют скорее о возможном “перераспределении” железа при ХСН из костного мозга в другие макрофагальные депо, где оно оказывается недоступным для эритропоэза даже при нормальном уровне сывороточного железа и ферритина, как это происходит при АХЗ [10].
Таким образом, абсолютный или относительный дефицит железа довольно часто встречается среди пациентов с ХСН и приводит к развитию анемического синдрома.
Нарушение функции почек
У больных ХСН нередко имеет место дисфункция почек с нарушением выработки ЭПО. Последний синтезируется преимущественно специализированными фибробластами, расположенными внутри коркового и мозгового слоев почек [11]. Основным сигналом для повышения выработки ЭПО служит снижение парциального давления кислорода. Как известно, почка весьма чувствительна к гипоксии, несмотря на то что получает около 25 % от сердечного выброса, а использует менее 10 % полученного кислорода. Согласно данным ряда авторов, уровень эндогенного ЭПО в крови у пациентов с ХСН достоверно выше, чем у здоровых людей, причем чем тяжелее степень ХСН, тем выше концентрация ЭПО [12, 13]. В то же время у пациентов с ХСН и наличием анемии чаще встречается низкая концентрация ЭПО. По-видимому, усиление выработки этого гормона почками в ответ на снижение перфузии при застойной ХСН непродолжительно [14]. При снижении фракции выброса на фоне застойной ХСН происходит уменьшение почечного кровотока [15], что в конечном итоге приводит к почечной дисфункции, которая обусловливает снижение выработки ЭПО с последующим развитием анемии [16].
Нарушения в ренин-ангиотензиновой системе
Анемия при ХСН может развиваться из-за нарушения в ренин-ангиотензиновой системе, а также вследствие приема ингибиторов ангиотензинпревращающего фермента (АПФ). Ренин-ангиотензиновая система играет важную роль в регуляции объема плазмы и числа эритроцитов. Увеличение концентрации ангиотензина II (АТ-II) в плазме приводит к изменению перитубулярного парциального давления кислорода [17]. Снижение парциального давления кислорода в перитубулярных фибробластах коркового вещества приводит к повышению концентрации активных форм кислорода внутри клетки, которые активируют фактор гипоксии HIF-1, увеличивая экспрессию гена ЭПО [18], т. е. АТ-II увеличивает секрецию ЭПО за счет эффектов снижения почечного кровотока и усиления реабсорбции в проксимальных канальцах. Имеются данные о том, что АТ-II оказывает прямое стимулирующее воздействие на эритроидный росток костного мозга [19]. Таким образом, применение ингибиторов АПФ и антагонистов рецепторов AT-II вызывает анемию путем снижения выработки ЭПО [20, 21]. В оригинальном исследовании А. Ishani и соавт. показано, что у пациентов с ХСН с нормальными показателями гематокрита на фоне приема эналаприла увеличивается частота выявления анемий в течение года. Тем не менее у пациентов, получавших эналаприл, выживаемость была выше по сравнению с пациентами, не получавшими данный препарат, даже при развитии анемического синдрома [20]. Это свидетельствует о том, что, несмотря на способность ингибиторов АПФ вызывать анемию, они остаются препаратами первого ряда при лечении ХСН.
Железоперераспределительный механизм (АХЗ)
При развитии ХСН отмечено нарастание концентрации фактора некроза опухоли α (ФНО-α), интерлейкина-6 (ИЛ-6) и других провоспалительных цитокинов [22], а также С-реактивного белка [23], что может приводить к уменьшению концентрации гемоглобина (Hb) [24]. Показано, что ИЛ-6 и ФНО-α ингибируют синтез ЭПО в почках путем активации гена GATA II и нуклеарного фактора В [25]. Это объясняет, почему у пациентов с ХСН снижается выработка эндогенного ЭПО после кратковременной стимуляции его синтеза. Кроме того, интерлейкины способны напрямую ингибировать эритроидный росток костного мозга [26], усугубляя анемию, хотя механизмы их действия остаются невыясненными. В экспериментах на крысах с индуцированной ХСН показано уменьшение числа клеток-предшественников эритроидного ростка и клеток, синтезирующих ЭПО [25].
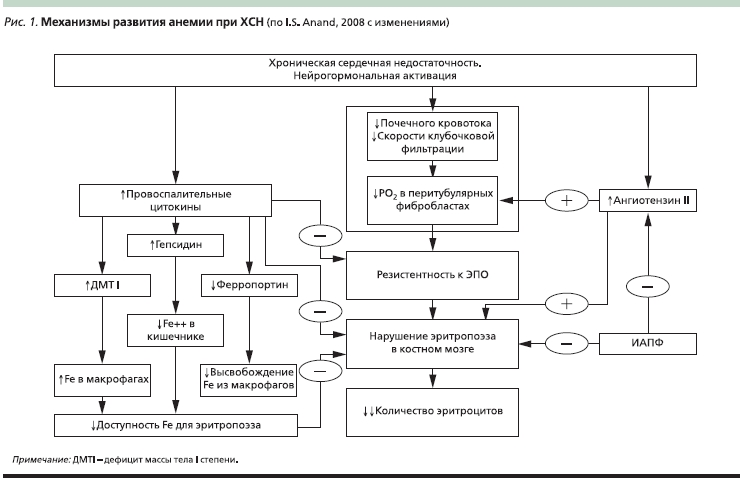
В клиническом исследовании С. Opasich и соавт. причина анемии среди 148 пациентов с ХСН была выявлена лишь у 43 %, причем только у 5 % больных была диагностирована ЖДА. У остальных 57 % пациентов верифицировать генез анемии не удалось. Следует подчеркнуть, что именно у данной категории больных были выявлены нарушения синтеза гема, а также низкий уровень эндогенного ЭПО и высокая активность провоспалительных цитокинов, несмотря на адекватные депо железа в организме [8]. Еще одним механизмом развития анемии в условиях высокой концентрации ИЛ-6 является увеличение синтеза гепсидина печенью, который в свою очередь уменьшает всасывание железа в кишечнике. Кроме того, ИЛ-6 ингибирует экспрессию белка ферропортина на мембране клеток-депо железа (энтероцитов, гепатоцитов, макрофагов). Этот белок отвечает за транспорт железа из клетки наружу, т. е. снижение его количества приводит к нарушению высвобождения железа из депо [10]. Аналогичный механизм развития анемии наблюдается у онкологических пациентов [27]. Таким образом, активация провоспалительных цитокинов – основной механизм развития АХЗ – во многом обусловливает развитие анемии у пациентов с ХСН. На рис. 1 представлены основные патогенетические механизмы анемии при ХСН.
Влияние анемии на прогноз больных ХСН
Большинство исследователей приходят к заключению о неблагоприятном влиянии анемического синдрома на прогноз у больных ХСН. Так, у пациентов с тяжелой ХСН снижение содержания Hb оказалось независимым предиктором смертности (относительный риск – 1,131, 95 % доверительный интервал – 1,045–1,224 для снижения уровня Hb на 1 г/дл) [28].
В исследовании W.H.W. Tang и соавт. пациенты с ХСН и анемией имели значительно худший 3-летний прогноз, чем лица с нормальным уровнем Hb: общая смертность составила 47 и 26 % соответственно (р < 0,0001). При дальнейшем мониторинге общая смертность за 3 года составила 58 % при персистирующей анемии и 45 % при вновь выявленной анемии против 31 % в отсутствие анемии [29]. Имеются данные о более частых повторных госпитализациях больных ХСН с наличием анемии по сравнению с пациентами, имеющими нормальные показатели Hb [30].
Анализ исследования SOLVD (Studies Of Left Ventricular Dysfunction) показал, что уровень гематокрита является независимым фактором смертности при ХСН, а по данным Фрамингемского исследования, анемия сама по себе расценена как независимый фактор риска для ХСН [31]. По-видимому, неблагоприятное влияние анемии на прогноз пациентов с ХСН обусловлен ремоделированием миокарда левого желудочка (ЛЖ) в условиях нейрогуморальной активации [32], перегрузки объемом и дополнительной гипоксии. В экспериментальных работах показано развитие гипертрофии миокарда, а в дальнейшем и дилатирование камер сердца при индуцированной тяжелой анемии у крыс [33]. В клинических исследованиях выявлена гипертрофия миокарда ЛЖ у пациентов с анемией на фоне хронических заболеваний почек, хотя не ясно, была ли она связана с анемическим синдромом или же с артериальной гипертензией [34]. Данных, указывающих на прямую взаимосвязь гипертрофии миокарда ЛЖ и анемии, в зарубежной литературе не встречается. В то же время в исследовании RENAISSANCE (Randomized Etanercept North American Strategy to Study Antagonism of Cytokines) показано, что увеличение уровня Нb на 10 г/л в течение 24 недель у больных хронической болезнью почек сопровождается снижением массы миокарда ЛЖ на 4,1 г/м² [35]. По данным Е.В. Гончаровой, у больных хронической ЖДА в 89,3 % случаев развивается кардиомиопатия, характеризующаяся на поздних стадиях заболевания глобальной диастолической дисфункцией обоих желудочков и развитием гипертрофии ЛЖ – в основном за счет межжелудочковой перегородки [36].
Нами проведено исследование влияния анемии на показатели центральной гемодинамики у больных ХСН [37]. Показано, что у большинства (91 %) пациентов с ХСН с наличием тяжелой анемии происходит инотропная стимуляция миокарда с развитием гиперкинетического типа кровообращения. Выявлена сильная обратная корреляционная связь (r = -0,78, р < 0,05) между величиной фракции выброса (ФВ) ЛЖ и уровнем Hb (рис. 2).

При этом оказалось, что увеличение ФВ и ударного объема происходило только вследствие изменения конечно-систолического объема без “привлечения” механизма Франка–Старлинга. В результате инотропной стимуляции миокарда на фоне анемической гипоксии ЛЖ способен развивать бoльшие напряжение и силу сокращения кардиомиоцитов при одной и той же величине конечного диастолического объема. Таким образом, тяжелая анемия вызывает у больных ХСН своеобразную эндогенную инотропную стимуляцию ЛЖ, что может оказывать негативное влияние на прогноз у данной категории пациентов. Такое предположение базируется на результатах многочисленных многоцентровых двойных слепых рандомизированных плацебо-контролируемых исследований, которые показали, что использование негликозидных инотропных стимуляторов увеличивает риск смерти больных ХСН. Так, в исследовании PROMISE показано, что применение инотропного препарата милринон у пациентов с ХСН вызвало рост общей смертности на 28 %, сердечно-сосудистой – на 34 %, а летальность в наиболее тяжелой группе больных застойной ХСН IV функционального класса по NYHA (New York Heart Association) возросла на 53 % [38].
Таким образом, коморбидность ХСН с анемией представляет собой важную клиническую проблему, требующую дальнейшего изучения и уточнения механизмов развития анемии при ХСН, влияния ее на прогноз больных, разработку оптимальных способов коррекции анемического синдрома.



{kind=link}