
Введение
Несмотря на огромный прорыв в лечении рака молочной железы (РМЖ), произошедший за последние десятилетия, мы продолжаем сталкиваться с неудовлетворительным ответом на лечение от многих пациентов. При этом более 2/3 случаев РМЖ относятся к люминальным подтипам (имеющим экспрессию ER – рецептор эстрогена и/или PR – рецептор прогестерона более чем в 1% клеток) и нуждаются на определенном этапе в эндокринотерапии, способствующей снижению частоты рецидивов и смертности от РМЖ [1].
Пролиферация и выживание как нормальной, так и раковой ткани молочной железы при этом происходит благодаря эстроген-индуцированной активации ядерных рецепторов ERα и ERβ. Рецептор эстрогена (ER) впервые был обнаружен в 1958 г. Е.В. Дженсеном. Последующие исследования показали, что эстроген (E2) вовлечен в патогенез РМЖ и способствует росту экспрессирующих ER клеток РМЖ.
ER при связывании с эстрогеном димеризуется и транслоцируется в ядро, где ER-димеры связывают коактиваторы с образованием транскрипционно активного ER-комплекса. Связанный с эстрогеном ER индуцирует прогрессирование клеточного цикла, в т.ч. за счет индукции экспрессии MYC и CCND1 (циклин D1) [2]. Стимулированный эстрогеном ER также усиливает митогенную передачу сигналов, регулируя транскрипцию нескольких факторов роста, важных для развития молочной железы, включая трансформирующий фактор роста α (TGFa), инсулиноподобный фактор роста 1 (IGF-1), амфирегулин и эпидермальный фактор роста (EGF) [3]. Супрессия эстрогена и антагонисты ER остаются основой лечения ER+-РМЖ в течение нескольких десятилетий. В качестве эндокринотерапии ЕR+-РМЖ одобрены селективные модуляторы ER (SERM), селективные ER-регуляторы (SERD) и ингибиторы ароматазы [4], а также ингибиторы циклинзависимых киназ 4 и 6 (CDK4/6) в сочетании с ингибиторами ароматазы или с SERD.
SERMs – это класс лекарств, действующих непосредственно на рецептор эстрогена, обладающих частичным агонистическим и антагонистическим потенциалом. Самым известным представителем данной группы считается тамоксифен. Этот препарат в течение последних четырех десятилетий повсеместно использовался в качестве препарата выбора пациентов с РМЖ. Впервые препарат был применен в качестве лечения метастатического заболевания; позже его использование было исследовано и в лечении раннего РМЖ, что привело к относительному снижению частоты рецидивов на 40–50% [5].
Механизм действия ингибиторов ароматазы заключается в снижении уровня эстрогена путем блокирования перехода андрогенов в эстрогены у женщин в менопаузе. Также ингибиторы ароматазы в комбинации с агонистами гонадотропин-рилизинг-гормона используются в эндокринотерапии пременопаузальных женщин группы высокого риска [6–8].
Предполагается, что SERD (например, фулвестрант) действуют главным образом путем индукции деградации белка ER или блокирования транскрипционной активности ER [9, 10]. Тем не менее недавнее исследование показывает, что фулвестрант и подобные антагонисты ER подавляют активность ER главным образом за счет нарушения внутриядерной подвижности ER [11].
С более широким применением эндокринотерапии возник феномен эндокринорезистентности. Так, почти у 20% пациентов с ранним ER+-РМЖ возникает рецидив заболевания [12]. Эндокринорезистентность неизбежно возникает при ER+метастатическом РМЖ (мРМЖ, рис. 1).
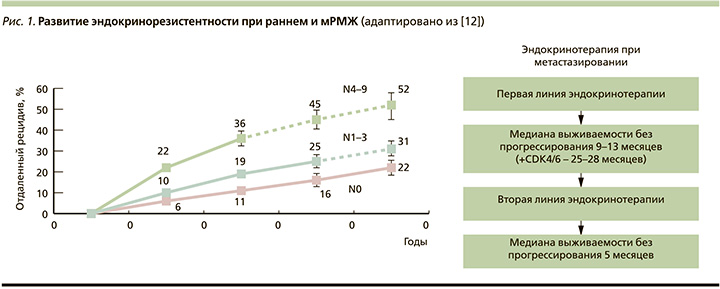
Согласно руководствам ESMO (European Society for Medical Oncology) и ASCO (American Society of Clinical Oncology), первичная эндокринорезистентность определяется как рецидив в течение первых 2 лет адъювантной гормонотерапии или прогрессирование заболевания в течение первых 6 месяцев гормонотерапии первой линии мРМЖ. Вторичная же эндокринорезистентность представляет собой рецидив после двух лет адъювантной гормонотерапии или в течение года после завершения адъювантного лечения, а также прогрессирование заболевания более чем через 6 месяцев после начала гормонотерапии при мРМЖ [13].
Первичная (врожденная) резистентность затрагивает меньше пациентов. У значительного же числа женщин развивается вторичная резистентность в ответ на эндокринотерапию. Изучается множество механизмов эндокринорезистентности, включая соматические альтерации, эпигенетические изменения и изменения в микроокружении опухоли.
Соматические изменения
1. Изменения в ER и ароматазе
Для многих методов лечения рака мутации в самой мишени, на которую направлено лекарственное средство, часто служат основным механизмом, посредством которого опухоль избегает ингибирования. ER является ядерным рецептором и состоит из двух подтипов: ERα и ERβ. ERβ оказывает противоположное действие по отношению к ERα и ингибирует стимулирующее действие E2 на пролиферацию клеток. Исследования показали, что подавление ERβ способствует прогрессированию опухоли и возможности выживания увеличиваются с ростом экспрессии ERβ [14, 15]. Тем не менее именно ERα преимущественно экспрессируется в опухолях молочной железы и считается важной мишенью эндокринотерапии.
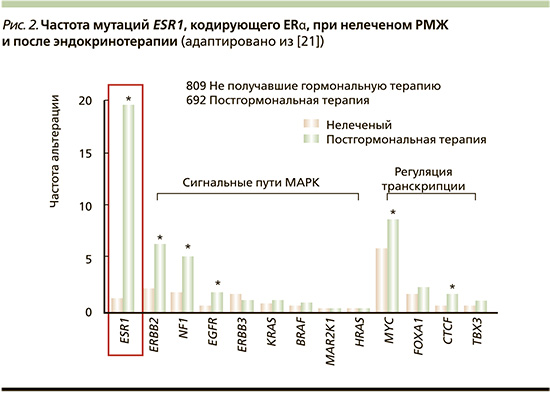
О точечных мутациях в ESR1, гене, кодирующем ERa, впервые было сообщено более двух десятилетий назад [16]. Однако только недавно признано, что приобретенные мутации в лиганд-связывающем домене (LBD) ESR1 служат частым фактором устойчивости при ER+-мРМЖ [17–20].
Эти мутации обнаруживаются почти при 20% рецидивов ER+-РМЖ, обычно приобретаемых после длительного лечения ингибиторами ароматазы или тамоксифеном [17, 21] (рис. 2). Распространенность мутаций ESR1 в циркулирующей опухолевой ДНК в аналогичных популяциях еще выше [22, 23]. Эти точечные мутации LBD (чаще всего у Y537 и D538) допускают гормон-независимую транскрипционную активность ER, что приводит к устойчивости к ингибиторам ароматазы и снижению чувствительности к тамоксифену и фулвестранту [24]. Мутации LBD снижают активность фулвестранта [25], поэтому в разработке находятся пероральные SERM или SERD, нацеленные как на мутантные ER, так и на ER дикого типа [26].
CYP19A1 – ген, кодирующий ароматазу, также изменяется в резистентных к ингибиторам ароматазы опухолях. L. Magnani et al. [27] сообщили, что амплификация CYP19A1 встречается среди 21,5% пациентов, у которых рецидив возник во время терапии ингибиторами ароматазы, но лишь менее чем у 2% пациентов с первичными опухолями. Амплификация CYP19A1 увеличивает активность ароматазы и приводит к привлечению ER к генам-мишеням в отсутствие экзогенного эстрогена, таким образом индуцируя устойчивость к подавлению эстрогена нестероидными ингибиторами ароматазы. CYP19A1-амплифицированные клетки оставались чувствительными к фулвестранту и стероидным ингибиторам, таким как экземестан [27]. В клинической практике для преодоления мутаций ESR1 был разработан класс препаратов SERD [28]. Включение CYP19A1 в целевые панели секвенирования может выявить подмножество резистентных к ингибиторам ароматазы опухолей, которые были бы кандидатами для лечения с помощью SERD.
В связи с возможной утратой экспрессии стероидных рецепторов пациентам с рецидивирующим заболеванием необходимо выполнять биопсию, чтобы подтвердить диагноз и измерить экспрессию биомаркеров для определения курса дальнейшей терапии. В эпоху точной медицины и таргетной терапии становится все более важным иметь свежие образцы ткани опухоли для определения наилучшего подхода.
2. Прогестероновый рецептор
При РМЖ PR также играет важную роль. PR регулируется ER и необходим для развития молочной железы. Хотя нормальные эпителиальные клетки молочной железы экспрессируют отдельные рецепторы (ER и PR) на определенных клетках, рецепторы коэкспрессируются в онкогенных клетках [29]. Данные различных исследований отражают важность PR, при этом опухоли ER+-РМЖ с PR-отрицательным статусом имеют худший, чем опухоли с PR+, результат [30]. Потеря PR приводит к активации и активизации пути PI3K [31]. Одно исследование продемонстрировало, что PR в комплексе с ER и PELP1 способствует регуляции E2-зависимых генов-мишеней ERα, связанных с пролиферацией клеток РМЖ и устойчивостью к тамоксифену [32].
3. Изменения в рецепторах тирозинкиназ
Давно известно, что амплификация HER2 (human epidermal growth factor receptor 2, ERBB2) снижает чувствительность к лечению антиэстрогенами в первую очередь посредством активации альтернативных путей (PI3K-AKT и MAPK) [33]. Следовательно, современный стандарт лечения ER+/HER2+-опухолей представляет собой комбинацию антиэстрогенов и ингибиторов HER2. HER2-активирующие мутации вовлечены как в первичную, так и в приобретенную резистентность к эндокринотерапии [34, 35] и обнаруживаются почти в 5% эндокринрезистентных, не-HER2-амплифицированных случаев мРМЖ [21]. Клетки ER+-РМЖ и ксенотрансплантаты, экспрессирующие активирующие мутации HER2, устойчивы к эстрогеновой депривации и фулвестранту, а также плохо реагируют на ингибитор тирозинкиназы нератиниб [34, 35]. Однако комбинированная блокада HER2 и ER показала синергичную активность как in vitro, так и in vivo и комбинация нератиниба с фулвестрантом продемонстрировала перспективные результаты в лечении ER+-мРМЖ, содержащего мутации HER2 [36].
Продолжаются клинические исследования, объединяющие ингибиторы FGFR и фулвестрант (а также палбоциклиб) в лечении ER+-мРМЖ (NCT03238196, NCT04024436). Гиперэкспрессия FGFR4 и мутации горячих точек связаны с эндокриннорезистентным дольковым мРМЖ [37]; комбинация эндокринотерапии с FGFR4-селективным ингибитором, таким как физогатиниб [38], считается возможной стратегией лечения этих пациентов.
4. Изменения пути PI3K
Соматические мутации PIK3CA встречаются примерно в 20–40% случаев раннего РМЖ, при этом чаще обнаруживаются при гормон-рецептор-позитивном (HR+) заболевании [39, 40]. Аберрантная активация пути PI3K способствует приобретенной устойчивости к cнижению уровня эстрогена в доклинических моделях [41, 42].
В исследовании SAFIR у 22% пациентов от общей популяции и у 28% пациентов с метастатическим HR+/HER2 РМЖ была обнаружена мутация PIK3CA, что аналогично показателям при раннем РМЖ [43, 44]. Интерес к PIK3CA-мутации недавно увеличился из-за публикации исследования SOLAR1, в котором продемонстрировано клинически значимое улучшение выживаемости без прогрессирования при применении α-селективного ингибитора PI3K. Алпелисиб, специфический ингибитор продукта PIK3CA, PI3Ka, был недавно одобрен в комбинации с фулвестрантом для лечения ER+-мРМЖ, содержащего мутации PIK3CA [45].
Ингибитор mTOR эверолимус, который блокирует критический сигнальный узел ниже PI3K, одобрен в комбинации с ингибиторами ароматазы для лечения мРМЖ, прогрессирующего на эндокринотерапии, независимо от мутационного статуса PIK3CA [46, 47]. Ингибитор AKT капивасертиб в сочетании с фулвестрантом также продемонстрировал предварительную эффективность при эндокринно-резистентном ER+- РМЖ [48, 49]. Эта комбинация может быть особенно эффективной при ER+-РМЖ с мутацией AKT1 [49, 50]. Мутации AKT1 и PTEN чаще встречаются при распространенном/метастатическом ER+-РМЖ [51, 52]. Приобретенные мутации горячих точек в PIK3CA были зарегистрированы после прогрессирования либо при терапии фулвестрантом, либо в комбинации его с ингибиторами CDK4/6 [53]. Важно отметить, что PIK3CA-мутантный рак, который прогрессировал в схемах, содержащих CDK4/6, оставался чувствительным к комбинации алпелисиба и фулвестранта [45].
5. Изменения пути MAPK
Компоненты сигнального пути MAPK, включая NF1, KRAS/NRAS/HRAS, BRAF и MAP2K1, часто являются мутированными при многих типах рака, но редко при первичном РМЖ. Однако мутации в этих генах, в частности ядерного фактора 1 (NF1), чаще встречаются при мРМЖ. Потеря NF1 приводит к конститутивной активности RAS. Кроме того, анализ циркулирующей опухолевой ДНК (ctDNA) резистентного к ингибиторам ароматазы мРМЖ выявил мутации KRAS, HRAS или NRAS у >15% пациентов [54].
6. Регуляторы генной экспрессии
P. Razavi et al. [21] обнаружили накопление изменений в регуляторах транскрипции MYC, FOXA1, CTCF и TBX3 при эндокриннорезистентном мРМЖ. Они были взаимоисключающими с изменениями в компонентах пути ESR1 или MAPK. Было обнаружено, что изменения в MYC, FOXA1 и CTCF либо существовали и ранее, либо приобретались после эндокринотерапии. Ранее признак активации Myc был связан со слабым ответом на тамоксифен со стороны пациентов [55]. CTCF служит репрессором транскрипции Myc [56]; предполагается, что мутации потери функции CTCF могут приводить к активизации Myc. Снижение экспрессии Myc с помощью ингибиторов BET может быть потенциальной стратегией для преодоления устойчивости в опухолях с изменениями MYC или CTCF [57, 58]. FOXA1 служит фактором, участвующим в ремоделировании хроматина, и было показано, что он взаимодействует с ER для индукции экспрессии генов [59]. Опухоли с мутациями FOXA1 могут все еще зависеть от экспрессии белка ER и, следовательно, оставаться чувствительными к SERD. Однако мутации горячей точки промотора в FOXA1, которые увеличивали экспрессию FOXA1, были связаны со сниженной чувствительностью к фулвестранту [60].
7. Гены репарации ДНК
Высокая мутационная нагрузка вызвана плохим прогнозом ER+-РМЖ [61]. S. Haricharan et al. [62] обнаружили, что мутации потери функции и/или низкая экспрессия генов системы репарации ошибок репликации MutL (MLH1/3, PMS1/2) были связаны с эндокринной резистентностью путем устранения CHK2-опосредованного ингибирования CDK4. Ингибиторы CDK4/6 остаются эффективными в MutL-дефектных клетках ER+-РМЖ.
Эпигенетические и негенетические механизмы
Эпигенетическое перепрограммирование
Структурные вариации и миссенс-мутации в генах, кодирующих ферменты, которые руководят эпигенетической регуляцией, часто участвуют в патогенезе РМЖ и устойчивости к эндокринотерапии. Накопление соматических мутаций в ремодуляторах хроматина, таких как гистонметилтрансферазы (KMT2B, KMT2D, KMT2E) и гистоновые деметилазы (KDM4A, KDM5B, KDM5C, KDM6A), отмечено, в частности, при люминальном подтипе РМЖ. Таким образом, более высокая активность KDM5 увеличивает гетерогенность опухолевых клеток и вероятность ранее существовавших клонов с первичной устойчивостью к эндокринотерапии. В соответствии с этим D.K. Patten et al. [63] показали, что под давлением отбора гормональной терапии эпигенетическое перепрограммирование способствует фенотипической гетерогенности и расширению эндокриннорезистентных клонов, недостаточно представленных во время начала лечения.
А. Stone et al. [64] идентифицировали гиперметилирование энхансеров ER-чувствительных генов как потенциальный механизм первичной и приобретенной эндокринной устойчивости. Таким образом, анализ метилом ДНК может служить эффективным инструментом для прогнозирования реакции ER+-РМЖ на ER-нацеленные препараты.
Активность абберантных кофакторов
Фактор FOXA1 открывает плотно упакованный хроматин, чтобы сделать его транскрипционно доступным для ER [65] и других факторов транскрипции. Амплификация/сверхэкспрессия гена FOXA1 в опухолях ER+ связана с низкой безрецидивной выживаемостью в ответ на тамоксифен [66].
Y. Johmura et al. [67] продемонстрировали, что при ER+-РМЖ потеря экспрессии белка F-box 22 (FBOX22) стабилизирует KDM4B, облегчая рекрутирование SRC и транскрипционную активность ER даже в присутствии SERM. Точно так же ко-репрессор ER NCoR важен для тамоксифена, выполняющего свою антагонистическую функцию. COPS5, который амплифицирован/сверхэкспрессируется в >85% опухолей, резистентных к тамоксифену, вызывает протеасомную деградацию NCoR и превращает тамоксифен в мощный агонист ER [68].
Эндокринная чувствительность при дольковом и протоковом РМЖ
Инвазивная лобулярная карцинома составляет до 15% инвазивного РМЖ и, как правило, является ER+. Инвазивная дольковая карцинома характеризуется потерей E-cadherin, что приводит к рыхлому фенотипу, связанному с лобулярным раком [69]. Ретроспективные исследования показали, что тамоксифен менее эффективен, чем летрозол, при терапии инвазивного лобулярного рака [70]. Доклинические исследования показали, что тамоксифен действует как агонист ER в клеточных линиях лобулярной карциномы и что ER-регулируемый транскриптόм отличается между клетками протокового и лобулярного РМЖ [71]. Соответственно, механизмы эндокринной резистентности могут различаться при инвазивном дольковом и протоковом раке. В эндокринорезистентность вовлечены FGFR1, WNT4 и липидный метаболизм, особенно в клеточных линиях долькового рака [71–73], хотя клиническое подтверждение этого отсутствует. C. Desmedt et al. [74] показали, что частота мутаций в PIK3CA, PTEN, AKT1, ERBB2, ARID1A и FOXA1, вовлеченных в эндокринорезистентность, выше в дольковых, чем в протоковых, раках. В их исследовании мутации в ERBB2 и AKT1 были связаны с повышенным риском раннего рецидива. Наконец, A. Pearson et al. [52] сообщили, что изменения потери функции NF1 встречались чаще при эндокринорезистентном метастатическом дольковом раке.
Опухолевое микроокружение
В эндокринорезистентность вовлечено несколько компонентов микроокружения опухоли, таких как гипоксия, опухоль-ассоциированные фибробласты, внеклеточный матрикс, экзосомы, а также клетки воспаления и иммунного ответа.
Гипоксия
Воздействие на индуцированный гипоксией переносчик аминокислот SNAT2 может повысить чувствительность клеток РМЖ к антиэстрогенному лечению [75].
Стромальные факторы
Опухоль-ассоциированные фибробласты играют ключевую роль в прогрессировании опухоли посредством ремоделирования экстрацелюллярного матрикса, секреции цитокинов и факторов роста, способствующих пролиферации и выживанию опухолевых клеток и модуляции иммунных клеток [76]. Опухоль-ассоциированные фибробласты могут секретировать цитокины или факторы роста, способствующие устойчивости к лекарственным препаратам. Например, H.M. Brechbuhl et al. [77] обнаружили, что кондиционированная среда из CD146-негативной подгруппы опухоль-ассоциированных фибробластов снижает экспрессию ER и чувствительность к тамоксифену, активирует тирозинкиназные рецепторы в клетках MCF7.
Внеклеточные везикулы могут переносить белки, ДНК и РНК между клетками и способствовать прогрессированию рака [78]. P. Sansone et al. [79] обнаружили внеклеточные везикулы, полученные из опухоль-ассоциированных фибробластов, содержащих митохондриальную ДНК, у пациентов с эндокринно-резистентным мРМЖ. Авторы показали, что внеклеточные везикулы, содержащие митохондриальную ДНК, способствуют уклонению от метаболического покоя в клетках, на которые воздействовал фулвестрант посредством восстановления окислительного фосфорилирования. Эти внеклеточные везикулы также способствовали самообновлению раковых стволовых клеток, способствуя устойчивости к фулвестранту.
Воспалительные и иммунные компоненты
Недавние исследования обнаружили вовлечение воспалительных цитокинов в эндокринорезистентность. Лечение антиэстрогенами индуцирует цитокин TGFb в клетках РМЖ, что приводит к устойчивости к антиэстрогенам и иммуносупрессии [79]. В неоадъювантном исследовании 112 пациентов, получавших ингибиторы ароматазы, сигнатура гена воспаления была самым сильным коррелятом плохого антипролиферативного ответа [80]. Фактически провоспалительные цитокины, такие как интерлейкин-1b и фактор некроза опухоли, стимулируют не зависимую от эстрогенов активацию транскрипции ER посредством IKKb-зависимого фосфорилирования ER Ser305, что приводит к эндокринорезистентности [81]. Следовательно, микроокружение опухоли напрямую передает сигнал ER и опосредует лиганд-независимую активность ER. Несмотря на эти новаторские исследования, роль воспалительных цитокинов в эндокринорезистентности остается недостаточно изученной областью.
Опухоли ER+ считаются иммунологически «холодными» из-за низкой инфильтрации опухоль-инфильтрирующими лимфоцитами [82]. Только около 20% ER+-клеток РМЖ экспрессируют лиганд-1 запрограммированной клеточной гибели (PD-L1), при этом монотерапия чекпоинт-ингибиторами показала ограниченную эффективность в опухолях, позитивных по PD-L1 и ER+ [83]. Показано, что ERα негативно регулирует экспрессию PD-L1 [84]. Следовательно, лечение антиэстрогенами может вызывать экспрессию PD-L1 и усиливать воздействие чекпоинт-ингибиторов. Клинические испытания, объединяющие ингибиторы контрольных точек и антиэстрогены в лечении ER+-мРМЖ, продолжаются.
Использование ингибиторов контрольных точек может быть особенно актуальным в ER+-опухолях люминального B подтипа, плохо реагирующих на ингибиторы ароматазы.
M. Anurag et al. [85] продемонстрировали, что высокая экспрессия компонентов иммунной контрольной точки IDO1, LAG3 и PD1 была связана с резистентной к ингибиторам ароматазы пролиферацией в опухолях люминального В-подтипа.
Использование ингибиторов CDK 4/6
Реактивация ER, опосредованная митогенным путем, включает CDK4/6-циклин D1-зависимую инактивацию Rb и дерепрессию факторов транскрипции E2F и, таким образом, чувствительна к ингибированию CDK4/6 [86]. Соответственно, добавление ингибиторов CDK4/6 (например, палбоциклиба, рибоциклиба) к антиэстрогенам заметно увеличивает выживаемость без прогрессирования по сравнению с терапией только антиэстрогенами пациентов с ER+-мРМЖ. Многие из описанных в данном обзоре механизмов резистентности могут быть нивелированы добавлением ингибиторов CDK4/6 к антиэстрогенам. Тем не менее они все еще могут оставаться актуальным фактором эндокринной резистентности при опухолях ранней стадии.
На чувствительность РМЖ к эндокринному лечению влияет активность как положительных, так и отрицательных регуляторов клеточного цикла. Исследования показали, что сверхэкспрессия положительных регуляторов клеточного цикла, таких как c-MYC, циклины E1 и D, способствует развитию эндокринной резистентности путем активации CDK [87, 88]. CDK4, активатор транскрипции E2F, служит модулятором не зависимой от E2 пролиферации клеток РМЖ, и ингибирование CDK4 приводит к ингибированию роста ER+-клеточных линий в отсутствие E2 [55]. Эти результаты подтверждают необходимость разработки ингибиторов CDK4 в качестве возможных средств для лечения РМЖ.
Идентификация биомаркеров может помочь выделить подгруппы пациентов, которые могли бы извлечь наибольшую пользу от терапии палбоциклибом, а также выявить механизмы резистентности, что приведет к рациональному отбору пациентов для комбинированной терапии с использованием ингибиторов CDK4/6.
N.C. Turner et al. [89] предоставили анализ генной экспрессии тканей РМЖ в исследовании PALOMA-3 и идентифицировали первый предиктивный маркер эффективности ингибиров CDK4/6, при этом низкая экспрессия мРНК CCNE1 была связана с большей эффективностью палбоциклиба. Источник биопсии опухоли оказал влияние на связь между экспрессией мРНК CCNE1 и эффективностью палбоциклиба. МРНК CCNE1 обладала высокой предиктивностью при биопсии из метастатического очага (n=142; р<0,001), но несущественной в образцах биопсии из первичного очага (n=159; р=0,09).
Единственным более значимым геном был нейромедин U, также вовлеченный в лекарственную устойчивость HER2-позитивного РМЖ, вызывая повышенные уровни трансформирующего фактора роста-1 [90] и расширяя фенотип стволовых клеток рака [91]. Отмечено, что более высокая экспрессия мРНК CDKN2D (p19) также связана с пониженной эффективностью комбинации с палбоциклибом, вероятно, как и экспрессия мРНК CDKN2C (p18). Оба гена принадлежат семейству INK4, регулирующему киназную активность CDK4/6.
Выводы
Масса исследований за последние несколько лет по многомерным аспектам эндокринорезистентности при мРМЖ позволяет предположить, что могут быть еще неопознанные факторы, способствующие эндокринной резистентности.
Нет сомнений в том, что ингибиторы CDK4/6 трансформировали лечение распространенного ER+-РМЖ. Комбинация ингибитора CDK4/6 с ингибиторами ароматазы или фулвестрантом в настоящее время служит стандартным лечением первой линии ER+-мРМЖ. Следовательно, основное внимание в исследованиях должно быть уделено предотвращению или преодолению резистентности к комбинации эндокринной терапии и ингибиторам CDK4/6. Также было показано, что некоторые из соматических изменений способствуют устойчивости к ингибиторам CDK4/6, включая амплификацию FGFR1, изменения PTEN и мутации ERBB2 [35, 93, 94]. В отличие от этого изменения RB1 и FAT1, по-видимому, связаны исключительно с устойчивостью к ингибиторам CDK4/6 и в меньшей степени к антиэстрогенам [53, 95]. Хотя большинство ранних стадий ER+-РМЖ чувствительны к эндокринотерапии, широкий спектр механизмов эндокринной резистентности создает барьер для лечения ER+-мРМЖ. Внутриопухолевая гетерогенность изменений резистентности представляет собой серьезную проблему при лечении эндокриннорезистентных видов мРМЖ [54] и других лекарственно-устойчивых форм рака [96].
Потенциальные стратегии улучшения показателей лечения ER+-РМЖ включают: 1) лечение наиболее эффективной терапией или комбинациями препаратов для максимизации эрадикации опухоли (т.е. эндокринная терапия+ингибиторы CDK4/6, ингибиторы PI3Ka и т.д. при опухолях ранней стадии); 2) отслеживание циркулирующей опухолевой ДНК для выявления приобретенной устойчивости до клинического прогрессирования и определение выявленных геномных изменений; 3) терапевтическое воздействие на спящие клетки ER+-РМЖ для предотвращения рецидива.


