
Введение
Атипичный гемолитико-уремический синдром (аГУС) представляет собой ультраредкое заболевание, проявляющееся микроангиопатической гемолитической анемией (МАГА), тромбоцитопенией, острым почечным повреждением (ОПП) и характеризующееся неблагоприятным прогнозом: 75% больных либо умирают в момент острого эпизода, либо демонстрируют быстрое развитие почечной недостаточности, достигающей степени терминальной хронической болезни почек (ХБП) в течение года от начала болезни [1–2]. В последнее десятилетие установлено, что генетические аномалии белков, регулирующих альтернативный путь комплемента (АПК), которые считались причиной аГУС, следует рассматривать как фактор, предрасполагающий к развитию болезни [3]. Значение остальных дефектов пока не определено.
Среди факторов, необходимых для реализации генетической предрасположенности, важное значение имеет беременность. По данным французских авторов, из 100 взрослых женщин с аГУС клинический дебют заболевания был связан с беременностью 21 пациентки, при этом манифестация заболевания в послеродовом периоде отмечена у 79% этих больных [4]. Результаты исследований, проведенных в последнее время, позволяют предполагать, что любые осложнения беременности, а тем более их сочетание, могут стать дополнительным комплемент-активирующим фактором, инициирующим развитие аГУС у женщин с генетическим дефектом в системе комплемента [5]. Однако генетический профиль при акушерском аГУС до настоящего времени подробно не исследовался.
Целью нашего исследования стало изучение клинических особенностей акушерского аГУС в сопоставлении с генетическим профилем системы комплемента.
Материал и методы
В исследование были включены 5 пациенток в возрасте от 20 до 33 лет (28,2±4,8 года) с акушерским аГУС. До развития острой акушерской ситуации все 5 женщин были здоровыми. Ни у одной из них не было в анамнезе эпизодов тромботической микроангиопатии (ТМА).
Диагноз аГУС был установлен на основании сочетания МАГА, тромбоцитопении и ОПП при исключении таких «неакушерских» причин развития острой ТМА, как тромботическая тромбоцитопеническая пурпура (ТТП) и катастрофический антифосфолипидный синдром (КАФС) [6].
После установления диагноза аГУС все женщины получали лечение комплемент-блокирующим препаратом Экулизумаб в/в сначала в индукционном режиме (4 недели по 900 мг препарата 1 раз в неделю), затем – в поддерживающем (начиная с пятой недели – по 1200 мг каждые 2 недели). Генетическое исследование выполнено всем пациенткам в разные сроки после постановки диагноза аГУС. Ко времени исследования все они продолжали лечение Экулизумабом.
Генетический анализ проведен методами секвенирования следующего поколения с использованием наборов для обогащения экзома Genotek Clinical Exome (Illumina Inc., США) и секвенирования ДНК производства Illumina (Illumina Inc., США). Биоинформатическая обработка данных произведена в соответствии с регуляциями ACMG (США). Референсная последовательность Human genome 19 (hg19) build 37.
Статистическая обработка выполнена с помощью пакета прикладных программ SPSS. При статистической обработке данных рассчитаны средние значения и стандартные отклонения.
Результаты
В табл. 1 приведена клинико-лабораторная характеристика пациенток с акушерским аГУС.
У 4 из 5 пациенток настоящая беременность была повторной, при этом предшествующие беременности протекали, физиологически завершившись срочными родами с рождением здоровых детей.
У всех пациенток аГУС развился после родоразрешения. Во всех случаях его развитию предшествовали в различных комбинациях дополнительные комплемент-активирующие состояния (КАС): диарея, пищевая токсикоинфекция, преэклампсия (ПЭ), ручное отделение плаценты, кровотечение (в результате операции кесарева сечения, ручного отделения плаценты, операции кесарева сечения и/или ампутации матки), сепсис (табл. 1). В каждом случае КАС могло быть несколько.
Все пациентки имели полный симптомокомплекс ТМА: резкое снижение уровня гемоглобина (55,4±19,1 г/л) (в отсутствие признаков кровотечения) и числа тромбоцитов (34,2±25,2 тыс. в мкл), выраженное повышение уровня ЛДГ (4546,6±4917,01 ЕД/л), подтверждающее наличие микроангиопатического гемолиза (МАГА).
Поражение почек во всех случаях было представлено ОПП с быстро нарастающим повышением уровня креатинина сыворотки (СКр) (539,2±295,6 мкмоль/л), олигурией или анурией, потребовавшими начала лечения гемодиализом в сроки от 2 до 5 суток от дебюта болезни.
У 4 из 5 пациенток отмечена артериальная гипертензия, хотя до развития заболевания ни у кого из них не регистрировались подъемы АД, в т.ч. и во время беременности. В среднем уровень АД составил 165/100 мм рт.ст. У всех наших пациенток ТМА носила системный характер с признаками поражения не только почек, но и печени, легких, а также ЦНС.
Поражение ЦНС было представлено тяжелой энцефалопатией, угнетением сознания вплоть до комы, развитием генерализованного судорожного синдрома. Поражение легких, проявлявшееся признаками отека легких, массивной двусторонней инфильтрацией легких, прогрессирующей дыхательной недостаточностью с потребностью в вентиляционной поддержке, в большинстве случаев трактовалось как острый респираторный дистресс-синдром (ОРДС).
Поражение печени было представлено повышением уровня трансаминаз и билирубина и развивалось уже в момент манифестации заболевания одновременно с признаками поражения почек или даже опережая их (у двух пациенток). Именно наличие в дебюте заболевания у всех пациенток признаков поражения печени с повышением уровня трансаминаз заставляло в первую очередь проводить дифференциальный диагноз с HELLP-синдромом. Однако своевременно начатая интенсивная терапия, включающая кортикостероиды, инфузии свежезамороженной плазмы (СЗП), хотя и способствовала значимому снижению уровня трансаминаз и билирубина, не привела, тем не менее, к улучшению состояния родильниц в течение 48–72 часов после родоразрешения. Напротив, у всех пациенток наблюдалось дальнейшее ухудшение состояния с развитием тяжелой полиорганной недостаточности. Именно прогрессирующая отрицательная динамика уже после родоразрешения позволила 3 из 5 пациенток своевременно диагностировать аГУС и начать патогенетическое лечение.
У одной родильницы аГУС осложнился сепсисом, что, безусловно, затруднило и отсрочило постановку диагноза, и, соответственно, начало лечения до 2 недель. Одной пациентке диагноз был установлен лишь спустя месяц от дебюта заболевания.
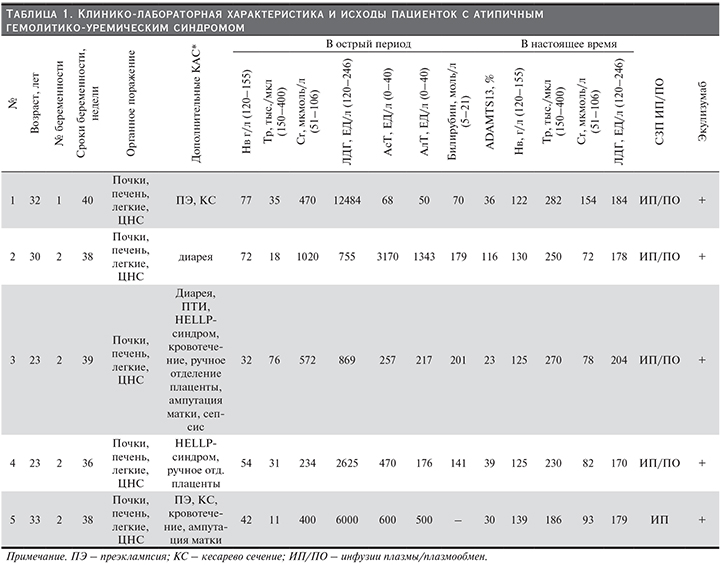
Для исключения другой первичной формы ТМА (ТТП) всем пациенткам определена активность ADAMTS13, показатели которой варьировались от 23 до 116%, составив в среднем 48,8% (референсные значения в РФ – 80–122%).
Все пациентки использовали (100%) СЗП, однако объемы ее значительно варьировались – у одной пациентки это была однократная инфузия СЗП (500 мл), в остальных случаях иело место сочетание инфузий СЗП (250–500 мл) и сеансов плазмафереза с объемом эксфузии и замещения не более 1,5 л. Ни в одном случае не было проведено ни одного сеанса полнообъемного плазмообмена (ПО). Помимо плазмотерапи пациенткам назначались низкомолекулярные гепарины и симптоматическая терапия (антибактериальная, дезинтоксикационная, антигипертензивная). Сразу после постановки диагноза аГУС всем пациенткам было начато лечение Экулизумабом: спустя 4 дня (№ 5), неделю (№ 2 и 4), 2 недели (№ 3) и месяц (№ 1) от дебюта болезни. В результате терапии Экулизумабом 4 из 5 пациенток, которым лечение было начато не позднее 2 недель, к концу периода индукции была достигнута клинико-лабораторная ремиссия аГУС с полным купированием всех проявлений ТМА, восстановлением гематологических показателей, функции почек и других жизненно важных органов. У пациентки № 1 (имеющей мутацию фактора H) с поздним началом терапии Экулизумабом (на 29-й день от дебюта аГУС) отмечена значительно более медленная динамика восстановления – значимый эффект появился только после 4-й инфузии: нормализовалось число тромбоцитов, улучшилось общее самочувствие, а полная гематологическая ремиссия была достигнута только к 6-му месяцу комплемент-блокирующей терапии, при этом функция почек так полностью и не восстановилась, хотя необходимости в диализной терапии больше не было. К настоящему времени терапия Экулизумабом продолжается более 2 лет, у больной сохраняется ХБП 3Б-стадии.
Пациентке № 5 (с мутацией фактора I) терапия Экулизумабом была прекращена после проведения индукционного курса, пациентке № 2 – через 1,5 года терапии, пациентке № 3 – спустя 10 месяцев от ее начала, причем в курсе поддерживающией терапии был перерыв сроком 1 месяц из-за сложностей с поставкой препарата. В настоящее время (спустя 1 год, 4 и 5 месяцев соответственно) после отмены препарата у всех женщин сохраняется ремиссия заболевания. Пациентки № 1 и 4 терапию Экулизумабом продолжают.
Все настоящие беременности закончились рождением живых детей, причем 4 из 5 новорожденных по массе тела при рождении соответствовали гестационному сроку и не имели признаков гипотрофии, 1 ребенок у пациентки № 1 с тяжелой пролонгированной в течение 4 недель ПЭ родился с признаками гипотрофии, однако в дальнейшем развивался без отставания.
Генетический профиль пациенток с акушерским АГУС
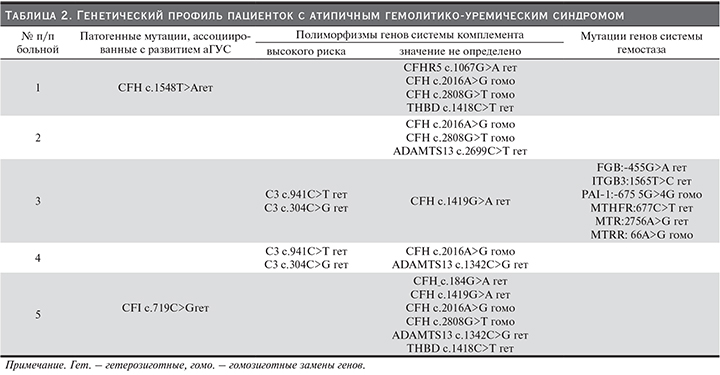
Всем женщинам было проведено генетическое исследование генов системы комплемента и гена ADAMTS13 (табл. 2). Мутации, ассоциированные с развитием аГУС, были обнаружены только у 2 пациенток: № 1 – мутация фактора Н (CHFc.1548T>A), № 5 – мутация фактора I (CHI c.719C>G). Еще у 2 пациенток (№ 3 и 4) были обнаружены мутации, описанные в литературе как полиморфизмы высокого риска (C3 c.941C>T; С3 c.304C>G)[7].Следует отметить, что полиморфизмы генов системы комплемента были выявлены у всех женщин (табл. 2), в т.ч. у 2 пациенток, имевших мутации, патогномоничные для аГУС. При этом наблюдалось совпадение одних и тех же полиморфизмов: фактора Н (CFH c.2016A>G и CFH c.2808G>T) – у пациенток № 1 (помимо мутации CHF), 2 и 5 (помимо мутации CHI); CFH c.1419G>A – № 3 и 5; С3 (C3 c.941C>T; С3 c.304C>G) – у пациенток № 3 и 4;THBD (c.1418C>T) – № 1 и 5.
Кроме того, у 2 женщин выявлена одна и та же мутация в гене ADAMTS13 (c.1342C>G) – № 4 и 5.
У одной пациентки (№ 2), у которой развитию аГУС после физиологически протекающей беременности предшествовала только кратковременная диарея накануне срочных родов, кроме генов комплемента дополнительно исследованы гены системы гемостаза. В отсутствие патогенных мутаций в генетическом профиле комплемента обнаружено 6 мутаций в генах, кодирующих белки свертывающей системы крови: 4 гетерозиготные мутации – FGB:-455G>A (фактор I, фибриноген), ITGB3:1565T>C (гликопротеин IIIа (GpIIIa), интегрин бета-3 (тромбоцитарный рецептор фибриногена), MTHFR:677C>T (метилентетрагидрофолатредуктаза), MTR:2756A>G (метионинсинтетаза) и 2 гомозиготные мутации: PAI-1:-675 5G>4G (ингибитор активатора плазминогена типа I) и MTRR:66A>G (метионинсинтетаза-редуктаза).
Обсуждение
Настоящая работа представляет собой первое в России исследование, в котором предпринята попытка определить генотипически фенотипические корреляции при акушерском аГУС.
Беременность служит одной из основных причин развития аГУС. По данным F. Fakhuri и соавт., из 100 взрослых женщин с аГУС клинический дебют заболевания был связан с беременностью 21 пациентки, из которых у 85% (18 из 21) были обнаружены дефекты в генах системы комплемента [4].Сегодня значимыми для развития аГУС признаны мутации фактора Н (CFH), мембранного кофакторного протеина (МСР), фактора I (CFI), фактора В (CFB), С3-компонента комплемента, из которых наиболее прогностически неблагоприятными считаются мутации в гене фактора Н [8]. Однако в последние годы в развитии аГУС стала обсуждаться помимо мутаций в генах системы комплемента роль однонуклеотидных полиморфизмов (SNPs) и их сочетаний [7, 9]. Результаты исследования генов системы комплемента на небольших выборках пациентов с аГУС из Китая (23 пациента) и Японии (10 пациентов) продемонстрировали, что треть пациентов с аГУС имели комбинированные мутации с недоказанным клиническим значением в различных генах CFH, С3, МСР (в одном случае выявлено 4 мутации в генах семьи CFH). Авторы предполагают, что синергичное взаимодействие множества полиморфизмов генов системы комплемента также способно приводить к развитию аГУС [7, 10].
В нашем исследовании, несмотря на небольшое число пациенток, у всех были выявлены дефекты генов системы комплемента различной значимости. Мутации, для которых доказана связь с развитием аГУС, согласно базе OMIM, были обнаружены только у 2 из 5 женщин, однако все пациентки имели полиморфные варианты генов системы комплемента с неясной на сегодняшний день клинической значимостью, но повторяющиеся в определенных комбинациях даже в нашей малочисленной группе. Так, замена c.1419G>A в гене CFH выявлена у 2 из 5 пациенток, составив 40%, в таком же проценте обнаружились замены c.1342C>Gв гене ADAMTS13 и c.1418C>T в генеTHBD. Кроме того, были обнаружены не только повторяющиеся единичные замены, но и сочетания вариантов генов: у 3 из 5 (60%) женщин – совпадающие полиморфизмы фактора Н (CFH c.2016A>G и CFH c.2808G>T), а у 2 из 5 (40%) – одинаковые полиморфизмы С3 (C3 c.941C>T; С3 c.304C>G). При этом аналогичные сочетания вариантов гена С3 (C3 c.941C>T; С3 c.304C>G) описаны и китайскими авторами, исследовавшими генетический профиль системы комплемента у 23 пациентов с аГУС и отнесшими эти изменения к группе полиморфизмов высокого риска для аГУС [7].
Кроме того, в последнее время обсуждается участие в патогенезе аГУС и генетических аномалий в системе гемостаза, в первую очередь мутаций генов ингибитора активатора плазминогена (PAI-1), тромбомодулина (THBD) и диацилглицеролкиназы эпсилон (DGKE) [11, 12]. Так, по данным литературы, при обследовании 36 больных спорадическим аГУС почти у 5% были дополнительно обнаружены мутации гена PAI-1 [11]. Предположение о дополнительном вкладе нарушений в системе гемостаза в развитие аГУС подтверждается примером одной из наших пациенток (№ 2), не имевшей значимых мутаций генов системы комплемента, но явившейся носителем множественных полиморфизмов генов системы гемостаза (всего 6), включая гомозиготную мутацию PAI-1 и 3 мутации ферментов фолатного цикла. Она единственная из наших пациенток не имела акушерских осложнений, но комбинированных генетических дефектов в системах свертывания крови и комплемента (хотя значение их и не определено), по-видимому, оказалось достаточным для развития практически фульминантного аГУС после возникновения диареи, предшествовавшей родам. Сочетание дефектов PAI-1 и фолатного цикла, обусловливающих соответственно неадекватный фибринолиз и дополнительное повреждение сосудистой стенки за счет гипергомоцистеинемии, можно рассматривать не только как дополнительный фактор, влияющий на выраженность ТМА и тяжесть течения болезни, но и как новое КАС, способное вызывать чрезмерную активацию АПК. Таким образом, мы полагаем, что результатом комбинации генетических аномалий системы комплемента и системы гемостаза с приобретенными факторами, активирующими комплемент (беременность и диарея), может стать развитие аГУС.
Следует отметить, что сама по себе физиологическая беременность, хотя и сопровождается умеренной активацией системы комплемента, обусловленной иммунологическим конфликтом между «полуаллотрансплантатом» – плодом и организмом матери, имеет как локальные механизмы защиты, ведущим из которых является экспрессия на поверхности трофобласта белков DAF (decay accelerating factor), MCP (membrane cofactor protein) и молекулы CD59, так и фетальные, наследуемые от родителей регуляторы активности системы комплемента [13–16]. Содружественная работа этих регуляторных факторов формирует многоуровневую антикомплементарную защиту, по-видимому, способную уравновесить даже незначительную активацию материнского комплемента, возможную при физиологическом течении процесса гестации. Данные анамнеза наших пациенток согласуются с этим предположением: у всех 4 повторнобеременных женщин, в т.ч. у пациентки, имевшей мутацию CFI, ассоциированную с развитием аГУС, предшествовавшие беременности протекали физиологически и закончились рождением здоровых детей.
Вероятно, угроза развития аГУС возрастает именно при отклонении в физиологическом течении беременности с возникновением акушерских осложнений и/или других факторов активации комплемента. Так, анализ течения настоящих беременностей показал, что у всех 5 женщин имелись дополнительные КАС (максимально 7), которые к тому же развились в короткий промежуток времени (не более 24–48 часов) непосредственно перед родами или как осложнение их и послеродового периода, следуя практически одно за другим (как пример: ПЭ–отслойка плаценты–кровотечение–оперативное родоразрешение), что, по-видимому, создавало суммационный эффект в отношении воздействия на систему комплемента и в конечном итоге приводило к взрыву комплементарной активности. Однако справиться с избыточной, неуправляемой активацией системы комплемента генетически дефектным белкам-регуляторам АПК в подобных ситуациях не удается, тем более что практически одновременно вместе с плацентой удаляются локальные регуляторы активации комплемента, что может провоцировать развитие акушерского аГУС у женщин с генетически обусловленным дефектом регуляции АПК. Для обозначения этой ситуации, как мы полагаем, уместно было бы использовать понятие «комплементарный шторм» по аналогии с тромботическим штормом, результатом которого является крайне быстрое генерализованное микроциркуляторное тромбообразование при катастрофическом АФС. Продолжая аналогию, а также учитывая темп развития полиорганной недостаточности вследствие ТМА при острых акушерских ситуациях, что иллюстрирует пример наших больных, можно предположить, что акушерский аГУС – это результат комплементарного шторма, вызванного сочетанным эффектом нескольких КАС, специфических для беременности.
Теория «комплементарного шторма» позволяет объяснить и быстрый положительный ответ на первую же инфузию комплемент-блокирующего препарата Экулизумаб у всех четырех повторнорожавших женщин. Очевидно, что действие дополнительных триггеров было достаточно мощным, чтобы вызвать неудержимую активацию АПК, но при этом слишком кратковременным для ее поддержания даже у женщин с дефектом генов системы комплемента. В таких случаях своевременное назначение Экулизумаба позволяет быстро и эффективно блокировать разрушительное действие комплемента. Это положение хорошо иллюстрирует пример пациентки с мутацией CFI, которой был проведен лишь индукционный курс Экулизумаба, приведший к быстрой (к концу 2-й недели) ремиссии заболевания с полным восстановлением гематологических и функциональных почечных показателей. Несмотря на то что больным аГУС, имеющим мутации CFH, CFI, C3, прекращать терапию Экулизумабом не рекомендуется из-за высокого риска рецидива болезни [17–19], наблюдение за пациенткой в течение года не выявило каких-либо клинико-лабораторных изменений, свидетельствоваших о возможном обострении. Кроме упомянутой пациентки после купирования острого эпизода ТМА и стабилизации состояния Экулизумаб был отменен еще двум женщинам, у которых тоже не возникло обострения болезни, несмотря на носительство нескольких полиморфизмов в генах белков-регуляторов АПК. В связи с этим есть основания полагать, что даже множественные дефекты в генах системы комплемента, которые проявляют свое патологическое действие только в экстремальной ситуации, по-видимому, могут «нейтрализовываться» в физио-
логическом состоянии другими, пока не определенными факторами. Наша гипотеза сочетается с данными T. Sahutoglu и соавт., описавших собственный опыт отмены Экулизумаба 1 пациенту с аГУС, проанализировавших подобный опыт еще 24 больным аГУС в 7 опубликованных работах и заключивших, что в случаях стойкой нормализации функции почек (как это имело место у наших пациенток) попытка отмены препарата оправданна [20]. При этом следует помнить, что пациенты с мутациями MCP, анти-CFH-антителами, не идентифицируемыми мутациями и мутацией CFI имеют низкий риск рецидива аГУС, а наличие мутации CFH представляет собой серьезную опасность рецидива ТМА после прекращения комплемент-блокирующей терапии [20]. Пример единственной нашей пациентки, имевшей прогностически неблагоприятную гетерозиготную мутацию CFH (CFH c.1548T>A), у которой в исходе острого эпизода ТМА, несмотря на продолжающуюся терапию Экулизумабом, развилась ХБП 3Б-стадии и сохраняется тяжелая трудноконтролируемая артериальная гипертония, подтверждает это заключение, поскольку даже случайное удлинение интервала между инфузиями препарата привело к быстрому нарастанию креатинина крови, вернувшегося к исходным значениям после нормализации режима введения. Очевидно, что в случаях отмены Экулизумаба пациенты должны находиться под постоянным наблюдением и мониторировать гематологические показатели, уровень СКр крови и анализ мочи, чтобы своевременно возобновить лечение Экулизумабом, если появятся признаки обострения аГУС. Наш, пусть пока и небольшой, опыт наблюдения за 3 пациентками после отмены препарата полностью подтверждает справедливость этой рекомендации.
Заключение
Таким образом, при акушерском аГУС, как и при аГУС в целом, мутации в генах системы комплемента лишь предрасполагают к развитию заболевания. Для реализации этой предрасположенности необходимо воздействие дополнительных комплемент-активирующих факторов, что подтверждает справедливость теории «множественных ударов» и в отношении аГУС. Для акушерского аГУС такими дополнительными комплемент-активирующими состояниями в первую очередь могут служить различные осложнения беременности и родоразрешения, а также генетические дефекты свертывающей системы крови. Любое отклонение в физиологическом течении беременности может вызывать «комплементарный шторм» с молниеносным развитием комплемент-опосредованной ТМА с генерализацией микроангиопатического процесса и быстрым формированием полиорганного поражения. Это обусловливает более тяжелое часто фульминантное течение акушерского аГУС, в отличие от течения аГУС в целом [1, 5].Своевременная диагностика аГУС и незамедлительное начало терапии Экулизумабом позволяют не только спасти жизнь пациенткам с аГУС, но и полностью восстановить их здоровье.
Конфликт интересов. Коротчаева Ю.В. является лектором образовательной программы компании «Алексион». Профессор Козловская Н.Л. является экспертом компании «Алексион». Остальные авторы конфликта интересов не имеют.


