
Синдром множественных эндокринных неоплазий (МЭН) – это группа генетически разнородных наследственных заболеваний, при которых выявляются опухоли или аденоматозные гиперплазии в двух и более эндокринных органах. Как правило, это нейроэндокринные опухоли (НЭО) – новообразования, происходящие из клеток APUD-системы. Эти клетки способны продуцировать нейротрансмиттеры, нейромодуляторы или нейропептиды, которые выделяются из секреторных гранул в ответ на внешние стимулы, а при опухолевой трансформации клеток их секреция становится нерегулируемой. К клеткам APUD-системы относятся островковые клетки поджелудочной железы (ПЖ), нейроэндокринные клетки респираторной системы и желудочно-кишечного тракта (ЖКТ), парафолликулярные клетки щитовидной железы (ЩЖ).
В зависимости от эмбриологического происхождения выделяют следующие группы клеток, относящихся к APUD-системе:
1. Эндокринные клетки нейроэктодермального происхождения, секретирующие серотонин, мелатонин, катехоламины.
2. Производные нервного гребешка, секретирующие адреналин, норадреналин, серотонин, кальцитонин.
3. Производные эндо- или мезодермы – гастроэнтеропанкреатические и эндокринные клетки респираторного и урогенитального трактов (G-, A-, D-, ES-, ECL-, S-эндокринные клетки). НЭО – гетерогенная группа различных по локализации, характеру роста и клинической симптоматике опухолей, происходящих из нейроэндокринных клеток и, соответственно, имеющих сходные цитологические характеристики.
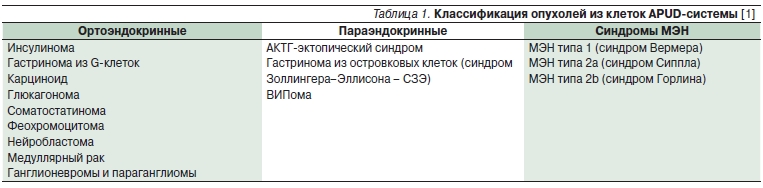
Таблица 1. Классификация опухолей из клеток APUD-системы [1].
Таблица 2. Характеристика синдромов МЭН [2, 3, 4].
Как видно из табл.1, синдромы МЭН входят в состав НЭО. Различают синдромы МЭН типов 1 и 2 (табл. 2).
МЭН-1 – редкий врожденный аутосомно-доминантный синдром, вызванный мутацией гена MEN1, локализованного на хромосоме 11q13 [5, 6]. Ген MEN1 имеет 10 экзонов (первый экзон некодирующий) и кодирует 610-аминокислотный белок менин. Хотя функции менина до сих пор полностью не изучены, предполагается его участие в репликации и репарации ДНК, транскрипции и модификации хроматина. В основном менин ведет себя как супрессор опухолевого роста [7–9], широко экспрессируемый в клетках эндокринных и неэндокринных тканей [10, 11]. У 40 % пациентов имеется сочетание опухолей ОЩЖ, гипофиза и ПЖ. Однако не у всех больных развиваются все типы опухолей. Диагноз синдрома МЭН-1 устанавливается при наличии опухолей минимум двух вышеперечисленных эндокринных желез, поражение которых наиболее характерно для синдрома МЭН-1, или при обнаружении хотя бы одной подобной опухоли у родственника пациента с синдромом МЭН-1 [11]. По данным Human gene mutation database (http://www.hgmd.cf.ac.uk/ac/index.php), в настоящее время описано более 540 различных геномных мутаций гена MEN1. Большинство из них является инактивирующим, приводящим к прекращению синтеза или изменению структуры менина и туморогенезу [12, 13]. За исключением группы геномных мутаций гена MEN1, найденных у пациентов с семейным изолированным первичным гиперпаратиреозом (ПГПТ), и мутаций MEN1-Burin в семьях с высокой частотой пролактином и низкой частотой гастрином, других генотип-фенотипических корреляций для синдрома МЭН-1 отмечено не было [12–16].
Клиническая характеристика синдрома МЭН-1 очень разнообразна даже у пациентов внутри одной семьи как по сочетанию пораженных органов, так и по возрасту начала заболевания, а также тяжести проявлений. В разных публикациях приводятся различные варианты частоты встречаемости тех или иных сочетаний поражения эндокринных желез и других органов при МЭН-1, что иногда зависит и от медицинского профиля научного учреждения, публикующего результаты своего исследования. Так, в нашем центре, который широко занимается проблемами опухолей гипофиза и ПГПТ, у 42 пациентов было выявлено сочетание ПГПТ с опухолями других эндокринных желез, наиболее часто – аденомами гипофиза (71 %). У 40 % больных имелись опухоли ПЖ, дополнительно у 30 % пациентов выявлены опухоли надпочечников. Опухоли другой локализации встречались намного реже.
У 17 пациентов на момент обследования было поражено более двух эндокринных желез одновременно, в т. ч. у 4 пациентов ПГПТ сочетался с опухолями и гипофиза, и ПЖ, и надпочечников.
По данным литературы (табл. 2) и результатам наших исследований, ПГПТ является наиболее ранним и частым клиническим проявлением синдрома с пенетрантностью до 100 % после 50 лет [17].
Энтеропанкреатические опухоли выявляются приблизительно у 60 % пациентов, причем на аутопсии эти опухоли встречаются в 100 % случаев [18]. В отличие от спорадических опухолей ПЖ МЭН-ассоциированные опухоли этого органа развиваются рано, почти всегда они мультифокальны и наблюдаются на всем протяжении железы. Клинические симптомы зависят от гормональной активности опухолей.
При гастриномах развивается СЗЭ, характеризующийся избыточной продукцией гастрина и гиперпродукцией соляной кислоты, что проявляется болями в животе, эзофагитом, пепсическими язвами и диареей [11]. Около четверти всех гастрином приходится на синдром МЭН-1 (18–26 %) [19]. Атипичное расположение язв, кровотечения из них и перфорации – характерные осложнения
гастрином. Для гастрином при МЭН-1 наиболее типична локализация в подслизистой оболочке двенадцатиперстной кишки (ДПК) и реже – в ПЖ. Минимум в 40 % случаев на момент обследования
уже имеются метастазы в лимфатические узлы и реже – в печень [20, 21].
Инсулиномы редко встречаются как первое проявление синдрома МЭН-1 и наблюдаются только у 10–30 % пациентов. До 90 % инсулином имеют диаметр менее 2 см. При семейной форме часто наблюдается мультигормональная секреция (до 50 % случаев). Клинически инсулиномы представлены триадой Уиппла (Whipple’s), признаками которой являются: гипогликемия натощак или вызванная физической нагрузкой; уровень глюкозы менее 50 мг/дл и исчезновение симптомов при введении глюкозы. В отличие от иных опухолей ПЖ инсулиномы часто бывают доброкачественными [22].
Другими активными опухолями являются редко встречающиеся энтеропанкреатические опухоли, секретирующие глюкагон, соматостатин или вазоактивный интестинальный пептид (ВИП) [23]. Крайне редко опухоли секретируют соматотропин-рилизинг-гормон, АКТГ и др. Для глюкагономы характерны признаки сахарного диабета, снижение массы тела, анемия, мигрирующая некролитическая эритема. Глюкагонома может обнаруживаться у пациентов при целенаправленном обследовании при подозрении на синдром МЭН или НЭО, когда она небольших размеров и протекает асимптомно. Симптоматические глюкагономы имеют тенденцию к избыточному росту и малигнизации [11].
ВИПомы, секретирующие ВИП, приводят к развитию синдрома тяжелой водянистой диареи, гипокалиемии и ахлоргидрии и также могут быть злокачественными [19].
Для соматостином характерны холелитиаз, диабет, стеаторея. Эти опухоли располагаются в ПЖ и ДПК. Они крайне редки и выявляются на этапе метастазирования без локализации первичной опухоли [24].
Третьим по распространенности характерным проявлением МЭН-1 являются аденомы гипофиза (20–60 %) [25, 26]. Наиболее часто они гормонально активны: пролактиномы и соматотропиномы, реже встречаются кортикотропиномы.
Также при МЭН-1 выявляют карциноиды производных передней кишки (тимус, бронхи, желудок, ПЖ, ДПК) [27]. Как правило, карциноид развивается после 50 лет и выявляется случайно, т. к. обычно имеет малосимптомное течение. Исключением является карциноид тимуса со склонностью к агрессивному течению и плохим прогнозом [28]. В редких случаях эти опухоли могут секретировать АКТГ и проявляться синдромом гиперкортицизма.
У пациентов с МЭН-1 нередко встречаются аденомы или гиперплазия коры надпочечников [11, 29] и опухолевые поражения ЩЖ [30]. Однако, возможно, в большей степени это связано с повышенной частотой проведения визуальных исследований у пациентов с МЭН, нежели с присущим данному заболеванию повышенным риском развития означенных нарушений [11].
Другие опухоли, такие как доброкачественные ангиофибромы, коллагеномы, подкожные или висцеральные липомы, маточные или пищеводные лейомиомы, менингиомы, спинномозговые эпендимомы [4], могут помочь в подтверждении синдрома МЭН-1 у пациентов с противоречивыми данными обследования, т. к. при этом заболевании они встречаются чаще, чем в основной популяции.
МЭН-1 можно заподозрить у пациентов моложе 30 лет с ПГПТ при вовлечении нескольких ОЩЖ, при рецидиве ПГПТ, с СЗЭ, мультифокальными опухолями ПЖ, двумя и более МЭН-ассоциированными опухолями [11, 31]. Показано, что при ПГПТ с умеренным повышенным или нормальным уровнем паратгормона у пациентов в возрасте до 50 лет вероятность наличия синдрома МЭН-1 увеличивается в 13 раз [32]. Таким пациентам необходимо назначение дополнительных лабораторных исследований и методов топической диагностики опухолей, характерных для МЭН-1, генетическое подтверждение синдрома МЭН-1. Определение нуклеотидной последовательности ДНК (из лейкоцитов периферической крови) проводится методом ПЦР с последующим секвенированием 2–10 экзонов и
прилежащих участков интронов гена MEN1.
Несмотря на то что распространенность этого заболевания невелика, выявление синдромальной формы опухолей эндокринных желез представляется важным при определении тактики лечения пациента, а также для своевременного обследования родственников. Для выявления МЭН-1 у родственников больного используют исследование выборочного фрагмента ДНК, мутация в котором является специфичной для этой семьи. Рутинное наблюдение пациентов без признаков болезни, но с генетически подтвержденным диагнозом включает сочетание биохимического и гормонального исследования крови и топическую диагностику опухолей ОЩЖ, гипофиза, из энтеропанкреатических эндокринных клеток, внекишечных карциноидов каждые 1–3 года [11].
В первую очередь в качестве лабораторного скрининга при МЭН-1 рекомендовано проводить измерение в крови уровней общего и ионизированного кальция, гастрина, хромогранина А (маркер НЭО), глюкозы, пролактина и инсулиноподобного фактора роста-1 [11].
Задачей скрининга является выявление нарушений на ранней стадии, когда опухоль легче всего поддается лечению и можно избежать последствий длительной гиперсекреции гормонов [33]. Относительно возраста начала проведения такого скрининга для выявления опухолей мнения расходятся. Некоторые специалисты рекомендуют начинать скрининг в раннем возрасте (с 5 лет). Другие советуют проводить скрининг с раннего пубертатного периода по причине редкости опасных для жизни осложнений МЭН-1 у маленьких детей [11].
Для раннего выявления ПГПТ в составе МЭН-1 необходимо у родственников больных мониторировать уровень кальция в крови и моче, а при тенденции к их повышению – определять уровень паратгормона и осуществлять дальнейшую диагностику. Как показывает наш опыт, обязательное определение уровня кальция в крови у пациентов с любыми аденомами гипофиза, установленным гиперкортицизмом, опухолями ПЖ и ЖКТ позволяет выявлять малосимптомные формы ПГПТ и в ряде случаев генетически подтверждать наличие синдрома МЭН-1.
Известно, что нейроэндокринные опухоли энтеропанкреатической области при МЭН-1 обычно невелики и часто гормонально неактивны, но могут увеличивать смертность этой категории пациентов в силу способности к малигнизации и метастазированию [3, 34].
В связи с этим особую важность приобретает их раннее выявление. Для диагностики энтеропанкреатических опухолей применяют совокупность визуализирующих методов, вклю-чающую сканирование с октреотидом, мультиспиральную компьютерную томографию (КТ) и эндоскопическое ультразвуковое исследование (УЗИ) [11, 19, 24], а также проводят исследование уровня хромогранина-А, повышение которого характерно для всех типов опухолей, в т. ч. гормонально неактивных [35].
Соматостатиновые рецепторы экспрессируются в большинстве нейроэндокринных опухолей [36]. Их присутствие имеет большое клиническое значение. Экспрессия рецепторов соматостатина и их количество в нейроэндокринных опухолях влияют на диагностику и эффективность консервативной терапии этих опухолей [37].
Наличие соматостатиновых рецепторов в опухоли обнаруживается при сцинтиграфии с меченым октреотидом, что позволяет почти в 85 % случаев локализовывать первичную опухоль и в 100 % – метастазы [37]. Этот метод наиболее информативен при диагностике гастрином, глюкагоном, карциноидных опухолей, реже – инсулином [38].
Определение уровня гастрина натощак необходимо для диагностики гастриномы, однако гипохлоргидрия и несиндромальный вариант язвенной болезни могут сопровождаться ложноположительными результатами [11]. Гипохлоргидрию можно исключать измерением базальной секреции соляной кислоты. Для подтверждения гастриномы определяют уровень гастрина после секретинового теста [39]. Часто гастриномы имеют небольшой размер и сочетаются с другими эндокринными опухолями, диффузно расположенными в ПЖ и ДПК [40]. При сравнении с другими методами топической диагностики, такими как УЗИ и КТ, сцинтиграфия с меченым ктреотидом позволяет значительно лучше визуализировать как первичные опухоли, так и метастазы гастрином [40]. Большинство исследователей предлагают использовать сочетание сцинтиграфии с октреотидом и эндоскопическое УЗИ как визуализирующие методы первой линии для определения локализации опухолей у пациентов с СЗЭ [37, 40].
В исследованиях, проведенных National Institutes of Health (NIH), показано, что чувствительность сцинтиграфии с меченным индием октреотидом составляет 92 % при выявлении печеночных метастаз гастриномы и 58 % для первичной опухоли любой локализации [19]. Чувствительность сцинтиграфии с октреотидом в отношении выявления гастрином, расположенных в ПЖ, остигает около 100 % [40].
Бета-клетки экспрессируют небольшое количество рецепторов к соматостатину, что ограничивает применение сцинтиграфии с октреотидом для визуализации инсулином. Чувствительность метода составляет 40–50 % [41, 42]. В таких случаях для уточнения локализации опухоли проводят КТ и эндоскопическое УЗИ. Кроме определения концентрации глюкозы натощак для выявления инсулиномы используют определение уровней инсулина, С-пептида и проведение пробы с голоданием [43]. Основными методами визуализации карциноида бронхов и тимуса является КТ, в то время как карциноиды желудка и ДПК могут быть обнаружены при эзофагогастродуоденоскопии или эндоскопическом УЗИ. При диагностике таких опухолей может быть также информативна сцинтиграфия с октреотидом (60–85 % наблюдений) [41, 42].
Наиболее сложным и дискутабельным остается вопрос выбора метода лечения опухолей при синдроме МЭН-1, что требует, как правило, индивидуального подхода в зависимости от возраста пациента и выраженности клинических проявлений.
По мнению большинства авторов, операцией выбора при ПГПТ у пациентов с МЭН-1 является тотальная паратиреоидэктомия и тимэктомия с аутотрансплантацией паратиреоидной ткани
и последующим длительным приемом аналогов витамина D [11, 44, 45]. Тотальная паратиреоидэктомия рекомендуется при МЭН-1 из-за частой встречаемости гиперплазий ОЩЖ и возможности рецидива ПГПТ. У пациентов старшего возраста, особенно при наличии противопоказаний к операции или рецидиве заболевания, в качестве консервативного лечения ПГПТ и его осложнений могут назначаться кальцимиметики [46]. У больных старшего возраста с мягкой формой ПГПТ, проявляющегося остеопенией или остеопорозом без переломов, показано лечение бисфосфонатами [47, 48] или кальцитонином [49].
Лечение аденомы гипофиза при МЭН-1 такое же, как и при спорадических случаях, и зависит от размера, а также активности аденомы. Возможно проведение аденомэктомии, медикаментозной (при пролактиномах и СТГ-секретирующих аденомах) и лучевой терапии [50]. Опухоли ПЖ маленькие и множественные, в силу этого встает вопрос о целесообразности их хирургического лечения. В одном ретроспективном исследовании проводился анализ результатов хирургического вмешательства при опухолях ПЖ менее 2 см в диаметре и было показано отсутствие преимуществ оперативного лечения по сравнению с консервативным [51]. Однако анализ данных другой когорты пациентов показал, что раннее выявление и оперативное удаление опухолей можно считать при МЭН-1 вполне оправданными [51]. Несмотря на то что в большинстве случаев (71 %) удаленные эндокринные опухоли ПЖ были гормонально неактивными, они часто оказывались злокачественными и метастазировали в лимфатические узлы и печень [24]. В ряде случаев метастазы выявляются уже после хирургического лечения [40].
При неоперабельных злокачественных нейроэндокринных опухолях, после хирургического удаления опухоли с высоким потенциалом злокачественности; в случае отсутствия четкой локализации опухоли при наличии симптомов ее гиперсекреции рекомендовано назначение биотерапии аналогами соматостатина [37].
В зависимости от степени злокачественности НЭО по современной классификации G0-G3 (по показателю злокачественного потенциала Ki67b и другим критериям) требуется назначение высоких доз аналогов соматостатина в качестве монотерапии или в сочетании с химиотерапией. В ряде случае биотерапия может дополняться иммунотерапией [37, 52] Наиболее часто с этой целью используется интерферон α [42].
Лечение инсулиномы хирургическое. Хотя в литературе содержится мало данных по этой проблеме, рациональным объемом операции принято считать дистальную панкреатэктомию с энуклеацией всех патологических образований, которые пальпируются или обнаруживаются при помощи интраоперационного УЗИ [24]. Дополнительно при гипогликемии может применяться диазоксид. Стрептозоцин и другие цитотоксические препараты могут уменьшить симптомы, сократив объем опухоли [52].
При выявлении гастриномы некоторые авторы предлагают более агрессивный подход, включающий раннее хирургическое вмешательство вне зависимости от размеров опухоли и уровня ее активности [33, 53, 54]. Другие рекомендуют лечение опухоли только при ее диаметре более 2,5–3,0 см [21, 55]. Основным обоснованием консервативного подхода является малый риск отдаленных метастазов при гастриноме менее 2,5–3,0 см в диаметре, в то время как резекция ПЖ чревата серьезными осложнениями [21, 34]. Объем операции сводится к дистальной панкреатэктомии, энуклеации видимых или пальпируемых образований в головке ПЖ. Кроме того, проводится регионарная лимфаденэктомия и дуоденотомия с местной резекцией всех опухолей в ДПК.
У пациентов с МЭН-1 при сочетании гастриномы с ПГПТ после паратиреоидэктомии как первого шага лечения при нормализации уровней паратгормона и кальция может происходить снижение концентрации гастрина и гиперпродукции соляной кислоты [11, 21], однако в большинстве случаев содержание гастрина остается повышенным (в несколько раз) вследствие его автономной продукции гастриномой. Ингибиторы протонной помпы оказываются эффективными в симптоматической терапии СЗЭ [11].
В то же время назначение аналогов соматостатина уменьшает гипергастринемию и подавляет рост эндокринных клеток ЖКТ у пациентов с СЗЭ [19]. Так как гипергастринемия является важным фактором в развитии гиперплазии энтерохромофиноподобных клеток, можно предположить, что уменьшение размеров опухоли связано с ингибированием продукции гастрина, а также с прямым действием аналогов соматостатина на энтерохромофиноподобные клетки, опосредованным специфическими соматостатиновыми рецепторами подтипа 2 [56]. При лечении трех пациентов с МЭН-1 в течение 6 месяцев аналогом соматостатина отмечено значительное уменьшение размеров гастринпродуцирующих опухолей с их полным исчезновением через год лечения [56].
По данным литературы, при 3-месячном лечении Сандостатин ЛАР карциноида кишки и ПЖ у 40–96 % больных исчезли клинические проявления опухоли и снизился уровень хромогранина А [38, 41]. Аналогичные результаты получены при лечении аналогами соматостатина ВИПом (65 %). ВИПома, так же как глюкагонома и соматостатинома, относится к числу редких опухолей ПЖ, с чем связано отсутствие точных рекомендаций по их лечению. При уточнении локализации опухоли выполняют дистальную панкреатэктомию, энуклеацию пальпируемых или видимых на УЗИ образований с региональной лимфаденэктомией [53], далее по показаниям применяют медикаментозное
лечение.
Таким образом, в настоящее время существуют широкие возможности точной диагностики всех проявлений синдрома МЭН-1, генетического подтверждения этого заболевания, что позволяет провести скрининговое обследование родственников пациентов с целью раннего выявления и лечения НЭО. Более того, на основе точного диагноза и современных методов хирургического и медикаментозного лечения значительно улучшается прогноз и увеличивается продолжительность жизни пациентов с разнообразными проявлениями синдрома МЭН.



{kind=link}
{kind=link}