
Согласно современным представлениям, хронический панкреатит (ХП) – это группа заболеваний поджелудочной железы (ПЖ) различной этиологии, характеризующаяся вариабельной воспалительной инфильтрацией паренхимы и протоков органа мезенхимальными клетками; итогом длительного персистирования такого воспаления является развитие необратимых изменений эктрацеллюлярного матрикса и фиброза с последующим нарушением функции органа [12].
В течение многих десятилетий ХП расценивался как достаточно однородное и, как правило, неизлечимое заболевание, наиболее часто встречающееся в районах с низким уровнем дохода на душу населения, который определенно влияет на качество продуктов питания и частоту злоупотребления алкоголем, а также табакокурения. Крупные клинические исследования последних десятилетий показали, что подобное представление является ошибочным и в определенной мере дискриминационным, а если глубже изучить историю панкреатологии – то даже неоснованным на реальных фактах [24]. Фиброз ПЖ с развитием функциональной панкреатической недостаточности при ХП, следуя современным представлениям, является исходом широкого диапазона экзогенных (экологических, инфекционных, нутритивных, токсических) воздействий и эндогенных (генетических, воспалительных) изменений, совокупность которых и приводит с вариабельным вкладом разных факторов к развитию заболевания у конкретного индивидуума [6]. Подобный подход к пониманию патогенеза ХП поддерживается большинством современных экспертов в области панкреатологии и отражен в современных классификационных системах этого заболевания [30].
Именно наличием генетических изменений и объясняется развитие многих случаев панкреатита и его осложнений. Следуя генетической теории развития и прогрессирования ХП, становится понятно, почему у одних лиц, длительное время злоупотребляющих алкоголем, развивается ХП, а у других – нет. Различные генные мутации и полиморфизмы, вероятно, определяют восприимчивость человека к развитию ХП, как правило, за счет ослабления протективных механизмов ПЖ [12]. Лавинообразное увеличение числа вновь открытых мутаций, описание которых ежегодно появляется в периодической печати, свидетельствует, что до сих пор известна только меньшая часть возможных генетических изменений, приводящих к развитию ХП. Поэтому канонической стала точка зрения, будто если диагноз “наследственный панкреатит” (НП) предполагается согласно особенностям клинического течения и семейному анамнезу, а генетический скрининг не выявляет конкретной мутации, то это не исключает данный диагноз [1].
Наследственный панкреатит: история изучения
Менее десятилетия назад мы определяли НП как редкую патологию ПЖ, клинически характеризующуюся повторными эпизодами острого панкреатита (ОП) в виде болевого абдоминального и диспепсического синдромов, постепенно увеличивающейся частотой и выраженностью рецидивов, нарастанием степени функциональной (экзокринной и/или эндокринной) панкреатической недостаточности, отягощенным семейным анамнезом, высоким риском рака ПЖ [1]. Сегодня мы вынуждены признать, что широкий спектр возможных ассоциаций гено-/ фенотипа НП колеблется от прямых аутосомных доминирующих черт заболевания с почти полной пенетрантностью (доминантные мутации гена катионического трипсиногена – PRSS1) через мягкие генетические факторы риска без признаков Менделевского наследования (мутации генов SPINK1 и CFTR) к очень тонким наследственным модификаторам болезни, которые могут быть идентифицированы только в очень крупных исследованиях (мутации генов химотрипсина С, анионического трипсиногена и др.). Таким образом, идентификация мутаций различных генов, в частности гена катионического (PRSS1) и анионического трипсиногенов (PRSS2), генов панкреатического секреторного ингибитора трипсина (SPINK1), кератинов и др., привела к изменению представлений о патофизиологии ХП, в некоторой степени определив значимость влияния факторов окружающей среды на степень пенетрантности НП, выраженность клинической симптоматики и возраст начала заболевания.
Переходя к новым открытиям в патофизиологии НП, нельзя не отметить ряд важнейших и любопытных фактов, ставших сейчас уже историей панкреатологии. НП был впервые описан в клинике Мейо в Миннесоте в 1952 г. M.W. Comfort и A.G. Steinberg, которые впервые предположили возможный генетический дефект в качестве причины ХП [9]. Наблюдая за семьей с 4 установленными и 2 предполагаемыми случаями рецидивирующего с раннего детства ХП, авторы отметили, что заболевание имеет аутосомно-доминантный тип наследования с неполной пенетрантностью в пределах 80 %. Продолжающиеся в течение последующего десятилетия в клинике Мейо исследования выявили еще четыре семьи с похожими изменениями. Любопытным стал факт обнаружения аминоацидурии у членов четырех семей из пяти, наблюдавшихся в 1962 г. [16]. В том же году появилась первая публикация, посвященная наблюдению семьи с НП за пределами клиники Мейо [10]. Авторы описали французскую семью из Нанта, характеризующуюся схожими с американскими семьями признаками. Следующее описание семьи с НП вновь последовало из-за океана [14], ознаменовав, пожалуй, закат “первой волны” научных работ, посвященных описанию отдельных семей. Оживленный интерес к проблеме НП в научных кругах поспособствовал тому, что в дальнейшем были опубликованы работы, включившие уже десятки семей и сотни наблюдений во всех регионах мира.
Безусловно на тот период патогенез заболевания не мог быть расшифрован. Именно в относительно более поздних работах с описанием серий случаев семейного панкреатита обнаруживались те или иные часто встречаемые находки и выдвигались гипотезы развития заболевания. Как правило, атаки панкреатита характеризовались повторными эпизодами выраженного болевого абдоминального синдрома в сочетании с повышением амилазы в сыворотке крови, а иногда и с лихорадкой.
В связи с отсутствием в те годы мощной диагностической базы, прежде всего методик визуализации, дифференциальный диагноз с семейной средиземноморской лихорадкой (периодическая болезнь) проводился только на основании имеющейся у ряда больных гиперамилаземии. Интересно, что и у здоровых родственников больных, страдавших болевыми приступами, были выявлены случаи гиперамилазурии. В дальнейшем было определено, что эта особенность характерна и для других семей, члены которых страдали рецидивирующим ХП с раннего возраста.
В 1972 г. R. McElroy и P. Christiansen описали семью, где 10 человек имели ярковыраженные клинические признаки ХП, а еще у 16 обследованных обнаружились отдельные его признаки, указывающие на возможность наличия заболевания. Кроме того, авторы отметили в данной семье высокую частоту развития тромбоза селезеночной и воротной вен [21], что косвенно свидетельствует о рецидивирующем течении НП –вероятно, в виде атак ОП.
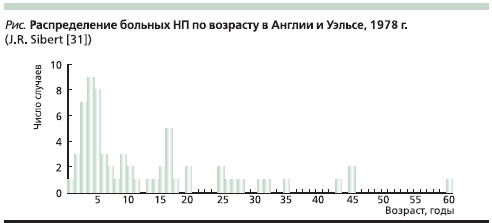
Наиболее крупная работа, посвященная изучению НП в эпоху отсутствия генетического тестирования, принадлежит J.R. Sibert и датируется 1978 г. Автором были выявлены 72 больных предположительно НП в 7 семьях в Англии и Уэльсе [31]. Заболевание наследовалось доминантно с вариабельной пенетрантностью, составившей в среднем, как и в пионерской работе M. Comfort и A. Steinberg [9], около 80 %. Необходимо отметить, что средний возраст первых проявлений заболевания был всего 13,6 года, что было сопоставимо с данными других исследований, хотя встречались и лица пожилого возраста (см. рисунок). В подавляющем большинстве случаев (81 %) клиническая манифестация НП развивалась до 20-летнего возраста. Важнейшее наблюдение автора заключается в идентификации двух пиков заболеваемости – в 5и 17-летнем возрасте [31], второй из которых, вероятно, указывал на манифестацию клинических проявлений рецидивирующего ОП у генетически предрасположенных лиц в связи с началом употребления алкоголя. В пяти обследованных семьях были выявлены лица с манифестацией заболевания и в детском, и во взрослом возрасте. Атаки панкреатита у большинства пациентов провоцировались стрессами, употреблением алкоголя и пищи с повышенным содержанием жиров либо комбинацией причин. С течением времени наблюдалось постепенное уменьшение частоты и тяжести болевых атак (что укладывается в классическую теорию соотношения некроза и фиброза). В 5,5 % случаев панкреатит характеризовался угрожающими жизни осложнениями. Признаки тяжелой экзокринной панкреатической недостаточности с синдромом мальабсорбции были зарегистрированы у 5,5 % больных, инкреторной недостаточности (панкреатогенного сахарного диабета) – у 12,5 %. Кальцификация и псевдокисты выявлены у 12,5 и 5,5 % больных соответственно. Тромбоз воротной вены был выявлен в 2 случаях из 72 и еще в 3 случаях – предположительно. Что примечательно: ни у одного из наблюдавшихся во всех семи семьях из разных регионов Великобритании не было отмечено аминоацидурии, имевшей место при первых описаниях больных НП в клинике Мейо.
Тот факт, что у больных НП при обзорной рентгенографии органов брюшной полости или на аутопсии очень часто находили кальцификаты в ПЖ, как правило, локализованные в ее протоках, а также иррегулярную дилатацию самих протоков, определил одну из наиболее ранних теорий этого заболевания – о наследственной патологии протоков [28]. Ведущую роль в этой теории отдавали повреждающему действию этанола, т. к. весьма сходные изменения протоков, включая кальцификацию и вирсунголитиаз, обнаружены и при НП, и при алкогольном ХП. Несостоятельность данной теории стала ясной после расшифровки генетической природы НП, хотя и спустя десятилетие ее авторы приводили случаи кальцифицирующего панкреатита с семейным анамнезом без фактов злоупотребления алкоголем [29]. Альтернативной теорией может служить предположение о наследственной изменяющейся вязкости панкреатического сока, способствующей преходящей панкреатической гипертензии и атаке ОП [22], которая частично подтвердилась в дальнейшем после открытия мутаций гена муковисцидоза (CFTR).
P.J. Robechek, описавший в 1967 г. пять случаев рецидивирующего с раннего детства панкреатита в одной семье, предполагал, что причиной НП являются передающаяся по наследству гипертрофия сфинктера Одди и наличие общей ампулы Фатерова сосочка для холедоха и главного панкреатического протока [26].
Наиболее поздней теорией, появившейся одновременно с первыми свидетельствами роли генетических изменений функций ряда белков ПЖ в развитии НП, стала гипотеза дефицита антиоксидантов [13], основанная на фактах снижения у больных с признаками НП активности компонентов антиоксидантной защиты, в т. ч. уровней витамина Е, селена и активности глутатионпероксидазы, а также повышения активности супероксиддисмутазы, что может приводить к накоплению свободных радикалов кислорода в клетках. Однако подобные изменения были обнаружены при многих этиологических формах ХП, характеризовавших скорее неконтролируемый воспалительный процесс, нежели специфику НП.
Итак, до середины 1990-х гг. диагноз НП был основан на наличии повторных атак ОП и/или персистирующих симптомов ХП, как правило, начинающихся в течение первых двух десятилетий жизни при наличии семейного анамнеза панкреатита по крайней мере у двух кровных родственников (трое и более больных панкреатитом в семье). Заболевание характеризовалось отсутствием половых различий, часто сопровождалось обнаружением внутрипротоковых кальцинированных конкрементов в отсутствие известных этиологических факторов, таких как алкоголь или холедохолитиаз.
К настоящему времени описано около сотни семей и тысячи пациентов, страдающих НП, причем генетические изменения идентифицированы практически у всех из них. Частота НП за последние три десятилетия возросла более чем в 4 раза. Большинство больных НП живут в странах Северной Америки и Европы, хотя семьи с НП сегодня описаны на всех континентах. Этому можно найти два объяснения: с одной стороны, в Северной Америке и Европе значительно лучше поставлена диагностика НП. С другой стороны, существуют данные о расовых особенностях заболеваемости НП. В частности, исследования американских ученных свидетельствует, что подавляющее большинство пациентов c НП относятся к белой расе, в то время как представители черной расы и коренные жители Америки болеют НП крайне редко. С середины 1990-х гг. несколько независимых групп исследователей начали проведение молекулярно-генетических исследований у больных НП с целью расшифровки патогенеза заболевания. Итоговым результатом стало практически одновременное обнаружение в 1996 г. в Европе и США двумя группами исследователей (D. Whitcomb и L. Bodic) мутации в 3-м экзоне гена катионического трипсиногена (PRSS1-ген) длинного плеча седьмой хромосомы (7q35) [19, 34]. Было установлено, что транзиция гуанина на аденин с изменением кодонов (CGC→CAC) приводит к замене аргинина на гистидин в положении 122 (R122H) аминокислотной последовательности фермента [35]. Вторая мутация в PRSS1-гене была впоследствии обнаружена во 2-м экзоне – N29I [15]. Эти две мутации (R122H и N29I) к настоящему времени идентифицированы в семьях с НП во многих странах, включая Францию, Германию, Испанию, Великобританию, Польшу, Японию, Китай, Южную Корею, США, и являются на данный момент самыми распространенными.
Наличие мутации R122H является причиной устойчивости трипсина к гидролизу, неконтролируемой каскадной активации трипсином других панкреатических проферментов и аутолиза ткани ПЖ. Как известно, в норме все проферменты активируются трипсином только в просвете двенадцатиперстной кишки после того, когда там под действием другой эндопептидазы – энтерокиназы кишечной каемки энтероцитов, происходит активация достаточного количества трипсиногена. Однако синтезируемый в ацинусах трипсиноген способен в незначительных количествах аутоактивироваться в ткани ПЖ, обеспечивая образование т. н. запального трипсина в панкреатическом секрете, вызывающего инициацию процессов пищеварения в двенадцатиперстной кишке на самых начальных этапах поступления панкреатического секрета в просвет последней [1].
Одним из механизмов, препятствующих в норме повреждению ПЖ, выступает панкреатический ингирбитор трипсина (ПИТ), который представляет собой специфический субстрат для трипсина, необратимо связывающий серин фермента с лизином своего активного центра и тем самым блокирующий около 20 % общего пула активированного трипсина в ткани ПЖ. Отношение синтезируемого ПИТ к трипсиногену составляет приблизительно 1 : 20. Когда уровень активности трипсина низкий, ПИТ предотвращает активацию трипсином других проэнзимов, однако количество трипсиногена значительно больше количества ПИТ. Поэтому в период интенсивной активации трипсиногена ПИТ не может выполнять свою защитную роль. В этих обстоятельствах трипсин и трипсиноподобные ферменты, как было сказано ранее, возвращаются в цепь гидролиза, объединяющую две шаровидные области трипсина в R122, что вызывает инактивацию фермента и остановку каскада. При мутации R122 и замене аргинина на гистидин в положении 122 трипсин и трипсиноподобные ферменты оказываются неспособными лизировать молекулы трипсиногена и трипсина, делая единственно возможным механизмом инактивации ПИТ. У больных НП ПИТ продолжает функционировать в обычном режиме, однако мощности его блокирующего эффекта оказывается явно недостаточной и при воздействии какого-либо провоцирующего фактора (например, алкоголя) может происходить чрезмерное превращение трипсиногена в трипсин, который не может быть инактивирован. В результате осуществится каскадная неконтролируемая активация панкреатических ферментов и аутодеструкция ПЖ, что в клиническом плане проявляется симптомокомплексом обострения ХП [1].
Механизм, посредством которого мутация N29I приводит к развитию НП, не совсем ясен. Предполагается, что эта мутация способствует аутоактивации трипсиногена, нарушая взаимодействие с ПИТ [15], либо препятствует инактивации трипсина, изменяя доступность начального участка гидролиза трипсина.
В течение предшествующего десятилетия были идентифицированы более 25 мутаций гена PRSS1, хотя большая часть из них были спорадическими семейными случаями. В дальнейших исследованиях четкой ассоциации между генотипом большинства выявленных наследственных изменений и фенотипом НП достоверно установлено не было, за исключением самых частых мутаций (N29I и R122H), которые и признаны ответственными за развитие НП. Тем не менее критерии для диагноза НП были пересмотрены, а диагностические критерии обновлены.
Исследования последних 5 лет окончательно показали, что НП с аутосомно-доминантным типом наследования определенно связан именно с R122Hи N29I-мутациями гена катионического трипсиногена и обнаруживается у 90 % больных с явными клиническими маркерами НП (ранний возраст клинической манифестации, высокая частота среди кровных родственников, наличие одного из родителей с анамнезом ХП и др.). В последние годы у больных наследственным или идиопатическим ХП описано еще более 10 мутаций и различных полиморфизмов этого гена, общая частота которых достигла 35. В связи с отсутствием четких фенотипических проявлений, характерных для НП, многие из этих редких мутаций были маркированы как условно ассоциированные с развитием ХП ввиду низкой пенетрантности. Действительно, большая часть описанных мутаций гена PRSS1 (R116C, R122C, N29T и т. д.) является спорадической, их роль в развитии ХП не является абсолютно установленной, а клинические исследования с целью установления их пенетрантности и фенотипа до сих пор не проведены. Поскольку НП – заболевание с аутосомно-доминантным типом наследования с неполной пенетрантностью, т. е. патология ПЖ возникает в результате Менделевского наследования одногенной мутации, знание пенетрантности чрезвычайно необходимо для определения генетического прогноза.
Среди прочих мутаций гена PRSS1 особый интерес вызывают мутации R122C, p.E79K и p.A16V.
Мутация R122C в гене PRSS1 была описана в отдельных небольших исследованиях, включивших только отдельные семьи. Наиболее крупная работа, датируемая 2009 г. [11], включает 22 испанские семьи. Авторами установлено, что мутация наследуется также аутосомно-доминантно с пенетрантностью 40,9 %. Это примерно соответствует данным, ранее полученным в небольших исследованиях, где пенетрантность R122C составила 33 [7], 50 [32] и 67 % [23]. Весьма любопытно, что в экспериментальной работе H. Archer и соавт. [5] на мышиной модели НП, воспроизведенной трансгенной экспрессией трипсиногена R122H, была сходная пенетрантность в пределах 40 %, поскольку выраженные изменения паренхимы ПЖ, типичные для ХП, развивались именно с такой частотой в течение первого года жизни трансгенных особей. Таким образом, относительно небольшая пенетрантность мутации R122C предполагает, что данное генетическое изменение – важный, но не облигатный фактор развития ХП.
Возвращаясь к наиболее крупной работе по изучению мутации R122C [11], следует отметить, что возраст клинической манифестации НП был выше, чем в среднем при мутации R122H, составив в среднем 23,5 года, а зарегистрированный возраст больных колебался в пределах 5–51 года. Болевой абдоминальный синдром встречался у 75 % пациентов с доказанным ХП, кальцификация выявлялась у 62,5 % больных, средний возраст которых составил 35,8 года (разброс – от 14 до 56 лет). Стеаторея выявлена в четверти случаев, сахарный диабет – значительно чаще: у 62,5 % пациентов. Рак ПЖ развился в 37,5 % случаев НП в возрасте от 59 до 70 лет [11]. В этой работе, а также в ряде предшествующих публикаций [7, 23, 32] отмечено, что при возникновении НП у молодых пациентов вследствие мутации R122C течение заболевания характеризуется отсутствием тяжелых атак ОП, преобладанием умеренных структурных изменений паренхимы и протоков, отсутствием тяжелой панкреатической недостаточности. Если диагноз НП устанавливался у лиц среднего и пожилого возраста, как правило, при обследовании регистрировались четкие лучевые признаки ХП. Эти данные укладываются в концепцию 30-летней давности, предложенную J.R. Sibert (1978) [31], и предполагают, что мутация R122C приводит к “мягким” и субклиническим изменениям структуры ПЖ. Причем такое относительно благоприятное течение НП существенно увеличивает анамнез заболевания, позволяя реализоваться накопленному предраковому потенциалу, о чем говорит высокая частота исхода этой формы НП в рак ПЖ [11]. Авторы показали, что риск развития рака ПЖ у больных НП, обусловленным мутацией R122C, в 757,6 раз выше, чем у лиц без генетических изменений.
Трипсиноген с мутацией Е79К не отличается от нормального ни по каталитической активности трипсина, ни по способности к аутолизу, ни по подавлению активности ингибитором SPINK1. Однако он активирует анионический трипсиноген PRSS2 по крайней мере в 2 раза сильнее, чем немутантный катионный трипсин. Поэтому Е79К может вызывать повышенную активацию трипсиногена не за счет аутоактивации, а вследствие трансактивации PRSS2. Однако клиническая значимость такой мутации остается неясной, т. к. она обнаружена и у здоровых лиц.
Мутация А16V не влияет на активацию трипсиногена, но в 4 раза повышает скорость активации трипсина in vitro. В отличие от R122Н и N29I, пенетрантность которых составляет 70–80 %, А16V обнаруживается почти исключительно у больных с семейным анамнезом панкреатита. Недавно у пяти семей с НП описана трипликация PRSS1и РRSS2-содержащего сегмента (около 605 пар нуклеотидов) [20]. Таким образом, помимо точечных мутаций в патогенезе панкреатита может играть роль повышение активности трипсина вследствие гендозозависимого эффекта. Значимость мутаций PRSS1 в патогенезе НП подтверждена недавним исследованием на трансгенных мышах с мутацией мышиного трипсиногена R122H. В ПЖ этих животных очень рано появились признаки поражения ацинусов, инфильтрация воспалительными клетками и усиленный ответ на индукцию панкреатита церулеином. Со временем у этих животных отмечены фиброз ПЖ и утрата дифференцировки ацинозных клеток [20].
Особенности клинической картины и диагностики наследственного панкреатита
НП чаще манифестирует в возрасте 3–5 лет. Начальные клинические проявления НП неспецифичны и могут быть сходными с клиникой ОП. Почти у 80 % пациентов в среднем наблюдается два обострения в год продолжительностью более двух дней. Частота атак ОП в ряде случаев имеет тенденцию к увеличению; по мере увеличиения длительности течения заболевания в первую декаду возрастает частота наиболее тяжелых форм. У 15–20 % больных достаточно быстро появляется выраженная стеаторея, регистрируются убедительные признаки ХП [1]. Для НП характерно увеличение длительности ремиссий с течением времени. На более поздних стадиях заболевания развиваются сахарный диабет (присоединяется через 8–10 лет у 20 % больных), тромбозы крупных вен (воротной, селезеночной, нижней полой), геморрагии. Гораздо чаще наблюдаются нарушение толерантности к углеводам, псевдокисты (5–10 %), кальцификация ПЖ (60–80 %), а также рак ПЖ [1].
Несмотря на то что заболевание является прогрессирующим, с частыми осложнениями, для НП характерна поздняя диагностика. Имеется второй пик выявляемости – в 18–25 лет, который в большинстве случаев совпадает с началом регулярного употребления алкоголя. Известны сообщения о более позднем наступлении клинической манифестации НП – в возрасте 30 лет. Идентификация НП традиционными методами бесполезна, поскольку его морфологических и биохимических маркеров не существует. Данные методик визуализации также неспецифичны. При обследовании каждого пациента необходимо проводить тщательный сбор анамнеза, учитывать наследственность, никотиново-алкогольную зависимость, потерю массы тела. Хотя НП характеризуется ранним началом (у 80 % больных приходится на возраст до 20 лет), но практически всегда – поздней диагностикой, образованием кальцификатов ПЖ и псевдокист.
Наследственный характер панкреатита можно предполагать при совокупности данных, полученных при выполнении ультразвукового исследования, компьютерной или магнитно-резонансной томографии, эндоскопической ретроградной холангиопанкреатографии. Для НП характерна выраженность структурных изменений ПЖ, не коррелирующая с возрастом больного – гиперэхогенность паренхимы, уменьшение железы в размерах, кальцификация паренхимы, вирсунголитиаз, внутрипротоковая гипертензия и др. Для окончательной верификации диагноза НП необходимо выявление мутаций гена катионического трипсиногена с помощью полимеразной цепной реакции.
Ранее считалось, что диагноз НП основывается прежде всего на исключении основных этиологических факторов, в частности злоупотребления алкоголем и наличия желчных камней, гиперлипидемии, в первую очередь I, II и IV типов, при которых панкреатит возникает в 15–40 % случаев гиперпаратиреоидизма, при котором у 10–15 % больных наблюдается поражение ПЖ, дефицита α1-антитрипсина или другого наследственного заболевания ПЖ. Однако неполная пенетрантность НП, наличие второго пика его выявляемости, по времени совпадающего с началом приема алкоголя, неритмичный характер обострений, предполагающий наличие провоцирующих факторов (погрешности в диете, прием токсичных лекарственных средств, обструктивный компонент) [1], в совокупности подтверждают экспериментальные данные об отсутствии облигатного развития НП у лиц с мутацией гена PRSS1, оставляя значимое место в развитии этого заболевания экзогенным факторам.
Один из главных рисков персистирующего НП – резкое повышение вероятности развития рака ПЖ. Частота регистрации рака ПЖ у больных НП колеблется в пределах 5 %. Кумулятивный риск развития рака ПЖ у больных ХП достигает 11 и 49 % у мужчин к возрасту 50 и 75 лет соответственно, а также 8 и 55 % – у женщин к аналогичным возрастным рубежам [25]. Этот вопрос изучался в двух независимых исследованиях в США и Европе, которые выявили повышение риска развития рака ПЖ у больных НП в 50–70 раз. Эти исследования проводились на основе баз данных Международной группы по изучению НП (International Hereditary Pancreatitis Study Group) и Европейского регистра НП и рака ПЖ (EUROPAC — European Registry of Hereditary Pancreatitis and Pancreatic Cancer). Риск рака ПЖ повышается начиная с 40 лет и достигает 40–70 % в возрасте 70 лет (особенно если НП прослеживается по мужской линии) [17]. Чаще всего рак развивается в семьях с мутациями R122H, N291 или же без мутаций, но с явным фенотипом НП. В то же время при генетических исследованиях больных НП изменений онкогенов или генов-супрессоров опухолевого роста не обнаружено. Это позволяет предположить, что причина высокой частоты возникновения рака ПЖ при НП заключается в более высоком уровне активности воспалительного процесса и большей его длительности. Это лишний довод в пользу того, что само хроническое воспаление является предрасполагающим фактором развития рака ПЖ. Вероятно, имеют значение длительность воспалительного процесса и отсутствие этиотропной терапии при НП.
Принципы терапии наследственного панкреатита
В настоящее время не существует специфической терапии НП. В случае обострения тактика идентична таковой при ХП – дезинтоксикация, купирование болевого синдрома, коррекция эндокринной недостаточности, нутритивная поддержка, антибиотикотерапия по показаниям.
Купирование болей является наиболее важной задачей лечения ХП и достигается при исключении употребления алкоголя, прекращении курения, использовании методики лечебного питания, применении фармакотерапии (ферментные препараты, блокаторы желудочной секреции, анальгетики и спазмолитики). Эффективность препаратов панкреатина объясняется тем фактом, что трипсин, входящий в их состав, оказывает ингибирующее действие на панкреатическую секрецию. Механизм действия заключается в инактивации холецистокининрилизинг-фактора с последующим снижением экспрессии холецистокинина, являющегося одним из основных стимуляторов секреции ПЖ. Для блокады панкреатической секреции содержание трипсина в просвете двенадцатиперстной кишки должно составлять 150–300 мг в течение часа. Содержание липазы, играющей не менее важную роль в ингибировании секреции ПЖ, для обеспечения гидролиза нейтрального жира должно быть не менее 20000 ЕД. Именно поэтому современные препараты панкреатина в виде минимикросфер (минимикросферы панкреатина, Креон) назначаются для купирования боли и в качестве заместительной терапии. При недостаточной эффективности ферментов для купирования болевого абдоминального синдрома наиболее часто используются ненаркотические анальгетики в больших дозах, в т. ч. и детьми, поскольку на данный момент равнозначной замены этим препаратам нет.
При выборе препарата для заместительной терапии необходимо назначать именно препараты 4-го поколения (энтеросолюбильные минимикросферы панкреатина), т. к. таблетки панкреатина из-за большого размера не проникают в двенадцатиперстную кишку одновременно с химусом и практически не принимают участия в гидролизе компонентов пищи. Другой причиной, объясняющей практическую непригодность таблеток пранкреатина, является низкое содержание липазы в одной таблетке, что определяет необходимость применения не менее пяти–семи таблеток на прием пищи, и это без учета того, что препарат частично инактивируется в желудке при “разваливании” крупной таблетки, так что потенциальная доза выше еще минимум в 2 раза. Поэтому только современные полиферментные препараты с активностью 25000–0000 ЕД липазы (Креон) способны полностью заместить экзокринную функцию ПЖ при ХП с внешнесекреторной недостаточностью. Их назначают пожизненно, при этом величина дозы зависит от соблюдения больным диеты. При выборе дозы панкреатина необходимо руководствоваться данными активности фекальной эластазы, коррекция дозы осуществляется по клиническим данным (купирование диареи и стеатореи, метеоризма, стабилизация и набор веса), лабораторным данным (снижение содержания жира в стуле, уменьшение объема фекалий, исчезновение нейтрального жира при микроскопии), результатам дыхательных изотопных исследований с мечеными триглицеридами или крахмалом. Только применение более современных препаратов, сочетающих кислотоустойчивость, одновременный с химусом пилоро-дуоденальный транзит, быструю активацию, высокое содержание протеаз, является залогом успешного лечения и улучшения качества жизни больных. Всеми проведенными исследованиями, опубликованными до настоящего времени, доказано, что наибольшей эффективностью при лечении панкреатической недостаточности характеризуются препараты 4-го поколения (Креон и аналоги), что в значительной степени определяет целесообразность использования Креона для купирования двух важнейших синдромов ХП – экзокринной панкреатической недостаточности и болевого абдоминального синдрома.
Доказано, что на фоне длительной (6 месяцев) высокодозовой монотерапии Креоном (100000–160000 ЕД липазы в сутки) достоверно реже возникали рецидивы болевого абдоминального и диспепсического синдромов, отмечена нормализация белкового обмена и нутритивного статуса с устранением дефицита массы тела, не было выявлено прогрессирования структурных изменений в ПЖ. Больные, постоянно принимавшие Креон, характеризовались наилучшими показателями качества жизни, не испытывали потребности в дополнительном сопутствующем лечении, среди них не было ни одного случая госпитализации по поводу обострения ХП [2]. Что интересно: у пациентов с ХП, длительное время получавших высокодозовую терапию Креоном, отмечено значительное и достоверное уменьшение экспрессии провоспалительных цитокинов с редукцией исходного дисбаланса между прои противовоспалительными цитокинами. Снижение экспрессии трансформирующего ростового фактора β на фоне применения Креона может свидетельствовать об уменьшении процессов фиброгенеза, поскольку известно, что в высоких концентрациях этот фактор стимулирует пролиферацию фибробластов и синтез коллагена, протеогликанов, гликозаминогликанов, фибронектина, тромбоспондина. Таким образом, длительный прием Креона может быть показан не только с заместительной целью при наличии внешнесекреторной недостаточности, но и с целью замедления прогрессирующего развития соединительной ткани в паренхиме ПЖ, в т. ч. и у больных без внешнесекреторной недостаточности, а также в качестве вторичной профилактики рака ПЖ [3].
Существуют единичные работы, посвященные поиску новых путей, направленных на купирование болевого абдоминального синдрома у больных НП, в первую очередь детей. Так, по данным G. Uomo и соавт., добавление антиоксидантной терапии (витамины А и Е, селен) позволяет уменьшать дозировки и длительность приема анальгетиков и уменьшать продолжительность болевого приступа [33]. Авторы провели экспериментальное исследование на трех молодых пациентах (кровных родственниках) с НП, продолжавшееся в течение 2 лет. Период наблюдения был разделен на четыре фрагмента по шесть месяцев каждый. В первом и третьем периодах пациенты получали по требованию только обезболивающие препараты внутрь; во втором и четвертом периодах был добавлен пероральный прием в день антиоксидантного витаминно-минерального комплекса: S-адеметионин – 800 мг (Гептрал), витамин C – 180 мг, витамин – E 30 мг, витамин А – 2,4 мг и селен – 75 мкг. Введение в схему терапии антиоксидантов привело к существенному сокращению (p < 0,05) частоты и выраженности болевого абдоминального синдрома в течение обоих периодов лечения, что привело к существенному снижению потребности в анальгетиках. По всей видимости, оксидативный стресс может быть одним из принципиальных факторов, потенцирующих болевой абдоминальный синдром у больных НП, а пероральный прием витаминно-минерального антиоксидантного комплекса эффективен для контроля выраженности боли у молодых пациентов, страдающих НП [33].
При развитии панкреатогенного сахарного диабета решается вопрос об инсулинотерапии.
Показанием к хирургическому вмешательству является наличие псевдокист, дуоденального или билиарного блока. Если существует настороженность в отношении рака ПЖ, то при выполнении эндоскопической ретроградной холангиопанкреатографии должна быть проведена биопсия эпителия панкреатического протока с дальнейшим цитологическим исследованием. В исключительных случаях в качестве профилактической меры рассматривается проведение панкреатэктомии, особенно у пациентов старше 30 лет и при наличии атипичных клеток [8].
Информация об авторах:
Кучерявый Юрий Александрович – кандидат медицинских наук, доцент кафедры пропедевтики
внутренних болезней и гастроэнтерологии ГОУ ВПО МГМСУ Минздравсоцразвития.
E-mail: proped@mail.ru;
Петрова Ника Валентиновна – доктор биологических наук, ведущий научный сотрудник,
лаборатория генетической эпидемиологии Медико-генетического научного центра РАМН.
E-mail: npetrova63@mail.ru;
Оганесян Татьяна Сергеевна – кандидат медицинских наук, ассистент кафедры пропедевтики
внутренних болезней и гастроэнтерологии ГОУ ВПО МГМСУ Минздравсоцразвития.
E-mail: atut77@rambler.ru;
Тибилова Залина Федоровна – аспирант кафедры пропедевтики внутренних
болезней и гастроэнтерологии ГОУ ВПО МГМСУ Минздравсоцразвития;
Смирнов Андрей Валерьевич – врач-гастроэнтеролог ФГЛПУ “Поликлиника № 2”
Минэкономразвития России


