
Введение
Рецептор эпидермального фактора роста (EGFR – epidermal growth factor receptor) – один из наиболее важных и наиболее изученных сигнальных путей, регулирующий рост, выживаемость, пролиферативную активность и дифференцировку клеток у млекопитающих [1]. Повышенная активность сигнального пути EGFR была обнаружена при многих видах опухолей, таких как рак поджелудочной железы, колоректальный рак, рак головы и шеи, немелкоклеточный рак легкого (НМРЛ). К этому могут приводить активирующие мутации в гене рецептора, увеличенное число копий этого гена или аутокринные петли (процесс, когда фактор роста, секретируемый клеткой, индуцирует синтез своего рецептора в той же клетке) [2].
Мутации гена EGFR встречаются примерно в 10% случаев в популяции больных НМРЛ Северной Америки и Западной Европы и в 30–50% случаев в странах Восточной Азии [3]. В российской популяции мутации гена EGFR встречаются среди 20% больных аденокарциномой [4]. Активирующие мутации гена EGFR находятся в 4 первых экзонах, кодирующих тирозинкиназный домен рецептора, – 18–21-й. Эти мутации очень разнообразны и включают точечные мутации, делеции и инсерции [3]. К наиболее распространенным мутациям относятся делеции в 19-м экзоне (45% случаев НМРЛ). Другая частая мутация – L858R в 21-м экзоне (40–45% случаев НМРЛ), изменяющая конформацию активационной петли тирозинкиназного домена. Замены нуклеотидов в 18-м экзоне (например, G719C или G719S) и инсерции в 20-м экзоне встречаются с одинаковой частотой – 5%. Редкие точечные мутации также могут встречаться ингибиторы тирозинкиназ в различных участках гена [3].
В настоящее время ингибиторы тирозинкиназы (ИТК)-EGFR служат стандартом терапии 1-й линии распространенного НМРЛ с наличием EGFR-мутации [5]. Однако у небольшой части пациентов нет ответа на таргетную терапию, что связано с наличием первичной резистентности к ИТК. У остальных пациентов после ответа на лечение неизбежно развивается прогрессирование заболевания, вызванное различными механизмами приобретенной резистентности.
В данной статье мы осветим основные механизмы, лежащие в основе первичной и приобретенной резистентности к ИТК-EGFR.
Первичная резистентность
Первичная резистентность имеется у 4–10% пациентов еще до начала таргетной терапии. У разных авторов встречается различное определение первичной резистентности от «отсутствия объективного ответа» до «наличия прогрессирования заболевания в качестве лучшего ответа опухоли на таргетную терапию» [6].
Хотя механизмы первичной резистентности еще не полностью изучены, их можно разделить на две группы:
- Зависящие от пациента (курение, полиморфизм гена BCL2L11 – BIM).
- Зависящие от опухоли (редкие EGFR-мутации, не отвечающие на терапию ИТК, молекулярные повреждения, связанные с мутацией EGFR, – активация PIK3CA).
Рассмотрим роль каждого из них.
Курение. Известно, что сигаретный дым вызывает активацию цитохрома CYP1A1, который является одним из цитохромов, вовлеченных в метаболизм ИТК-EGFR [7] и высвобождение реактивных компонентов оксидативного стресса, и запускает аутофосфорилирование, приводящее к уменьшению подавления «дикого» и «мутированного» EGFR даже в присутствии ИТК [8, 9].
Полиморфизм гена BCL2L11 (BIM). BIM – это белок из семейства BCL-2, который требуется для апоптоза, вызванного некоторыми видами ИТК, включая ИТК-EGFR [10–12]. In vitro-ингибирование экспрессии BIM вызывало резистентность к ИТК-EGFR.
Редкие мутации в гене EGFR. Не все мутации в гене EGFR имеют схожую чувствительность к ИТК. Так, хотя во всех исследованиях подтверждена чувствительность к ИТК для двух наиболее частых мутаций (делеции в 19-м экзоне del19ex и мутация L858R в 21-м экзоне), более редкие мутации в гене EGFR продемонстрировали устойчивость к данной терапии. Так, например, инсерции в 20-м экзоне встречаются с частотой 5–10%, характеризуются высокой вариабельностью, вызывают активацию рецептора EGFR, но в отличие от драйверных мутаций del19ex и L858R демонстрируют низкую аффинность к ИТК и являются таким образом причиной первичной резистентности [13].
Мутация Т790М, служащая превалирующей причиной приобретенной резистентности, редко, но может выявляться у первичных пациентов в небольшом количестве случаев. Наличие мутации Т790М в таких случаях обычно ассоциировано с наличием активирующей мутации и связано с меньшей частотой и меньшей продолжительностью ответа на ИТК-EGFR [14].
Молекулярные повреждения, связанные с мутацией в гене EGFR
Мутации в генах, вовлеченных во внутриклеточные пути, связанные с передачей сигнала от активированного EGFR, также могут приводить к запуску каскада внутриклеточных биохимических процессов, активации клеточного роста и пролиферации независимо от активности рецептора, таким образом приводя к первичной резистентности. Одним из таких механизмов является активация PIK3CA [15]. Соматические мутации PIK3CA встречаются примерно в 1–3% случаев НМРЛ [16].

Приобретенная резистентность
Приобретенная, или вторичная, резистентность развивается в клетках опухоли в ответ на проводимую терапию после объективного ответа или длительной стабилизации заболевания. D. Jackman и совт. предложили детальные критерии приобретенной резистентности к ИТК-EGFR [17]:
- Предшествующая терапия ИТК (в монорежиме).
- Наличие хотя бы одного условия:
- мутации гена EGFR, предсказывающей чувствительность к лекарственной терапии или клинический эффект от проведенного лечения (полный ответ/частичный ответ или стабилизация 6 месяцев и более);
- прогрессирования в ходе продолжавшейся в течение последних 30 дней терапии ИТК;
- отсутствия промежуточной системной терапии в период после окончания ИТК и перед началом последующей схемы лечения.
Приобретенная резистентность развивается у всех пациентов в ходе таргетной терапии ИТК-EGFR и может быть вызвана различными механизмами:
- модификацией EGFR, вызванной вторичной мутацией;
- включением обходных и нижележащих сигнальных путей;
- трансформацией фенотипа опухоли.
Модификация EGFR, вызванная вторичной мутацией
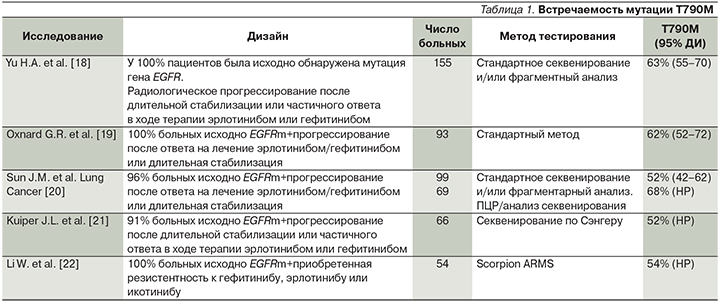
Наиболее частым механизмом вторичной резистентности является развитие мутации T790M в 20-м экзоне гена EGFR. Она составляет от 52 до 63% от всех случаев вторичной резистентности (табл. 1) [18–22].
Мутация Т790М вызывает вторичные изменения в структуре рецептора эпидермального фактора роста, что приводит к затруднению соединения молекулы ИТК 1-го поколения с рецептором и обеспечивает устойчивость опухолевой клетки к действию препарата [23–24]. Кроме того, приобретенная резистентность к ИТК может развиваться из-за возникновения мутаций в нижележащих сигнальных путях (RAS, RAF, MEK) или из-за активации обходных путей (MET, FGFR, HER2 и т.д.), а также в результате фенотипической трансформации опухоли в мелкоклеточный рак [25].
Согласно клиническим рекомендациям Всеобщей национальной онкологической сети (NCCN), обоснованно проведение биопсии при подтверждении прогрессирования заболевания на фоне терапии ИТК-EGFR пациентов с наличием активирующих мутаций в гене EGFR перед сменой терапии для идентификации механизма приобретенной резистентности. Определение механизма приобретенной резистентности к ИТК-EGFR может помочь подобрать последующую терапию [26]. Клинические рекомендации Европейского общества медицинской онкологии (ESMO – European Society for Medical Oncology) также предписывают проведение биопсии пациентам на момент прогрессирования на фоне терапии ИТК-EGFR для определения мутации Т790М в гене EGFR или другого альтернативного механизма резистентности [27].
Возможность выполнения и клиническая значимость проведения биопсии при прогрессировании НМРЛ были оценены в нескольких исследованиях (табл. 2) [18, 28–31].
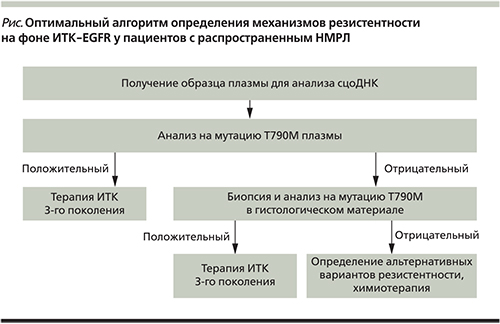
Определение мутации Т790М возможно также и в плазме. Исследование сцоДНК плазмы обладает рядом преимуществ перед биопсией: минимальная инвазивность, возможность получения материала в любое время (до, во время, после лечения), потенциальная возможность исследовать ДНК из всех клонов опухоли при условии ее гетерогенности [32]. Но исследование сцоДНК плазмы также обладает и рядом недостатков по сравнению с исследованием опухолевой ткани: в зависимости от времени, локализации опухоли, наличия некроза уровень циркулирующей опухолевой ДНК может быть низким или неопределимым, кроме того, на ранних стадиях заболевания или при ограниченном метастазировании число копий циркулирующей опухолевой ДНК может быть низким [32].
При сравнении результатов молекулярно-генетического тестирования плазмы и гистологического материала необходимо учитывать такие показатели, как чувствительность и специфичность метода. Чувствительность – показатель частоты ложноотрицательных результатов, а специфичность – показатель частоты ложноположительных результатов анализа. G. Oxnard и соавт. [33] оценили молекулярно-генетическое тестирование сцоДНК плазмы для выявления мутации Т790М при прогрессировании на фоне терапии ИТК-EGFR в исследовании AURA I.
Среди результатов 216 пациентов, имевших как гистологические образцы, так и образцы плазмы, чувствительность составила 70,3%, специфичность – 69%. Чтобы оценить, был ли 31% случаев (18 пациентов) с положительным статусом мутации Т790М в плазме и отрицательным в биопсийном материале ложноположительным, исследователи проанализировали эти образцы с помощью альтернативных методов и подтвердили наличие этой мутации в плазме 14 из 18 пациентов. Таким образом, положительный статус мутации служит не результатом низкой специфичности метода, а следствием гетерогенности опухоли. Чувствительность определения мутации Т790М в плазме пациентов в объединенном анализе исследований II фазы AURA и AURA2 составила 61,4%, специфичность при определении мутации Т790М составила 78,6% [13]. Эти результаты позволяют считать, что риск ложноположительных результатов невысок [34].
Мы также провели оценку плазмы как материала для определения активирующих мутаций EGFR и мутации Т790М у 35 пациентов, получавших гефитиниб в 1-й или 2-й линиях терапии. Для активирующих мутаций проведено сравнение частоты выявления с гистологическим материалом (чувствительность составила 83,3%, специфичность – 100%). Частота мутации Т790М составила 57,9% (в 36,8% случаев выявлена только Т790М, в 21,1% случаев – Т790М вместе с активирующей мутацией EGFR) [35]. Наши собственные данные сопоставимы с результатами международных исследований.
Учитывая вышеизложенные данные о возможностях и ограничениях как биопсии при прогрессировании на фоне первой линии терапии ИТК, так и анализа сцоДНК плазмы, возможный алгоритм диагностики может быть двухэтапным (представлен на рисунке).
Включение обходных и нижележащих сигнальных путей
Помимо развития вторичных мутаций EGFR опухолевая клетка может сформировать резистентность за счет активации внутриклеточных сигнальных путей в обход заблокированного рецептора EGFR, например, путем амплификации МЕТ, МАРК или НЕR2. Данные механизмы встречаются с частотой 5–12% и могут развиваться как совместно, так и независимо от статуса мутации Т790М [36].
Еще одним вариантом развития резистентности к ИТК помимо активации альтернативных путей может быть таковая эффекторных молекул на пути передачи сигнала от EGFR. Таким образом, даже при заблокированном и неактивном рецепторе происходит запуск каскада биохимических реакций, который поддерживает пролиферацию клеток [36]. Примером таких внутриклеточных каскадов служит передача сигнала RAS-RAF-MEK-ERK. Мутация BRAF может вызывать активацию данного сигнального пути и быть причиной вторичной резистентности. Данный механизм встречается с частотой около 1–2%. Другим примером служат сигнальный каскад PI3K-AKT-mTOR и мутация PIK3CA, которая вызывает его активацию, а также развитие не только первичной, но и приобретенной резистентности [36].
Трансформация фенотипа опухоли
Примерно у 3% пациентов может происходить перерождение опухоли в мелкоклеточный гистологический вариант. Механизмы, лежащие в основе такой трансформации, пока не изучены, но примечательно, что в таком случае опухоль демонстрирует высокую чувствительность к стандартным режимам химиотерапии [36].
Обсуждение
Накопленные данные о молекулярных механизмах опухолевого развития позволяют более точно подобрать вариант терапии 1-й линии для пациентов с распространенным НМРЛ. А понимание механизмов резистентности к ИТК может дать клиницисту дополнительные возможности для эффективной терапии второй и поздних линий, а также уточнить прогноз заболевания. С учетом полученных данных необходимо внедрять в клиническую практику гистологическую и молекулярно-генетическую диагностику перед назначением 2-й линии терапии при прогрессировании на фоне ИТК. Использование жидкостной биопсии может дать возможность ряду больных определить наиболее частые механизмы резистентности без использования инвазивных процедур, а остальным больным необходимо рассматривать возможность проведения биопсии для получения опухолевого материала и проведения гистологического и молекулярно-генетического анализа.


