
Печеночная энцефалопатия (ПЭ) – это синдром, объединяющий комплекс потенциально обратимых неврологических и психоэмоциональных нарушений, возникающих в результате острых или хронических заболеваний печени (с явлениями острой или хронической печеночноклеточной недостаточности соответственно) и/или портосистемного шунтирования крови [1]. В клинической практике наиболее часто явления ПЭ наблюдаются среди больных циррозом печени различной этиологии (вирусной, аутоиммунной, алкогольной и др.), будучи проявлением хронической печеночноклеточной недотаточности и нередко дополняясь ПЭ шунтового происхождения в связи с формированием выраженных портокавальных коллатералей [2].
Механизм развития печеночной энцефалопатии
Наиболее полно объясняющей механизмы ПЭ при циррозе печени является “теория глии”, согласно которой эндогенные нейротоксины и аминокислотный дисбаланс, возникающие в результате печеночноклеточной недостаточности и/или портосистемного шунтирования крови, вызывают отек и функциональные нарушения астроглии [3]. Вслед за этим развиваются повышение проницаемости гематоэнцефалического барьера, изменение активности ионных каналов, нарушение процессов нейротрансмиссии и обеспечения нейронов макроэргическими соединениями, что и приводит к клиническим симптомам ПЭ [4].
Среди эндогенных нейротоксинов ведущее место отводится аммиаку, баланс которого строго контролируется в организме [5]. В физиологических условиях наибольшее количество аммиака образуется в толстой кишке при гидролизе кишечной флорой белка и мочевины и в скелетной мускулатуре при физической нагрузке. Детоксикация аммиака в организме осуществляется по двум механизмам: биосинтез мочевины в печени в орнитиновом цикле и образование глутамина. Орнитиновый цикл – основной механизм связывания аммиака. В противоположность синтезу глутамина обезвреживание аммиака в орнитиновом цикле является окончательным, поскольку мочевина представляет собой продукт, выделяющийся почками [6].
Значение аммиака в патогенезе ПЭ подтверждается усилением симптомов ПЭ при приеме аммиакпродуцирующих веществ (например, белка) и исчезновением или уменьшением при назначении безбелковой диеты. Однако отсутствует четкая взаимосвязь между концентрацией аммиака в сыворотке крови и выраженностью симптомов ПЭ.
Кроме аммиака к группе эндогенных нейротоксинов относятся также меркаптаны, коротко- и среднецепочечные жирные кислоты, фенолы [7]. Другой причиной нарушения функции глиальных клеток является аминокислотный дисбаланс, лежащий в основе биосинтеза “ложных” нейротрансмиттеров, обладающих угнетающим действием на клетки головного мозга [8]. В патогенезе ПЭ также имеет большое значение повышение концентрации серотонина и γ-аминомасляной кислоты, количества рецепторов к ним в головном мозге.
Суммируя представленные данные, можно утверждать, что патогенез ПЭ является комплексным и включает действие нескольких факторов: эндогенных нейротоксинов (в первую очередь аммиака), аминокислотного дисбаланса, изменения в функционировании нейротрансмиттеров и их рецепторов (рис. 1).

Клиническая картина печеночной энцефалопатии
Проявления ПЭ варьируются от субклинических, выявляемых лишь специальными психометрическими тестами, до глубокой печеночной комы, являющейся наиболее частой причиной смерти при циррозе печени [9]. Большинство проявлений ПЭ обратимо при их лечении, однако у некоторых больных она прогрессирует с развитием деменции, спастического парапареза, церебральной дегенерации и экстрапирамидных двигательных нарушений, обусловленных органическими нарушениями центральной нервной системы. В таком случае следует говорить о необратимой ПЭ, которая может частично регрессировать после ортотопической трансплантации печени.
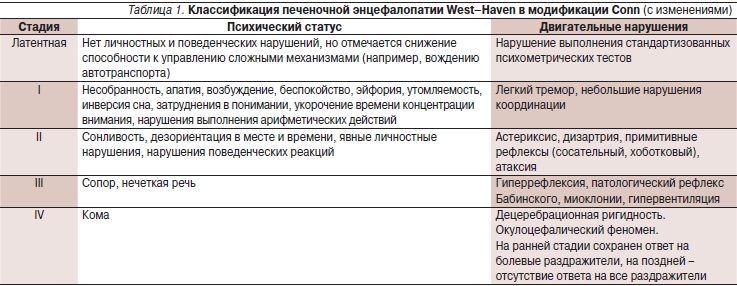
Клиническая диагностика ПЭ включает оценку нейропсихических симптомов: изменений интеллекта, поведения, нейромышечных нарушений, однако основной критерий диагностики и подразделения ПЭ на стадии – это изменение сознания (табл. 1). К ранним признакам изменения сознания относятся уменьшение числа спонтанных движений, фиксированный взгляд, заторможенность и апатия, краткость ответов. Дальнейшее ухудшение сознания ведет к тому, что больной реагирует только на интенсивные стимулы, затем развивается кома. Расстройства интеллекта характеризуются неспособностью больных повторять элементарные действия; нарушается почерк, страдает речь. Характерным неврологическим проявлением ПЭ является “хлопающий” тремор (астериксис) [10].
В настоящее время особое внимание уделяют латентной ПЭ, поскольку, клинически не проявляясь, данное состояние характеризуется снижением быстроты познавательной деятельности и точности тонкой моторики, что может реализовываться в невозможность обеспечения должного контроля, например, при управлении сложными механизмами (автотранспортом). Частота латентной ПЭ достигает 80 % среди больных компенсированным циррозом печени, для выявления которой используют психометрическое тестирование. Психометрические тесты также позволяют объективизировать нарушения функции центральной нервной системы при ПЭ I и II стадий. Выделяют тесты, направленные на определение быстроты познавательной деятельности (тест связи чисел – см. рис. 2 и тест число–символ), тесты для определения быстроты и точности тонкой моторики (тест линий и тест обведения пунктирных фигур). Достаточно простым в применении является тест складывания фигур, суть которого состоит в том, что больного просят сложить простые узоры (пятиконечную звезду, квадрат, домик и т. д.) из спичек или специальных палочек. У больных с латентной формой ПЭ в значительной мере страдает оптикопространственная деятельность, одним из вариантов нарушения которой и является конструктивная апраксия, проявляющаяся неспособностью скопировать простой узор из спичек или специальных палочек. Описанный выше тест характеризует как гностические возможности пациента, заключающиеся в узнавании пространственной фигуры, так и конструктивные – воспроизведение предложенной формы. Конструктивная апраксия выявляется также в нарушении почерка [11]. Для выявления этих нарушений целесообразно рекомендовать больному ежедневное ведение дневника, в котором он может отмечать диурез, массу тела и характеризовать свое самочувствие.

Правильно интерпретировать признаки поражения центральной нервной системы в пользу ПЭ помогают симптомы, свидетельствующие о печеночноклеточной недостаточности, одним из проявлений которой, собственно, и является ПЭ [12]. Принципиально важен факт возникновения симптомов ПЭ на фоне нарушения функции печени (в рамках хронической печеночноклеточной недостаточности – табл. 2).

Диагностика печеночной энцефалопатии
Клиническое подозрение на наличие ПЭ может быть подтверждено определением концентрации аммиака в сыворотке крови, однако необходимо помнить, что отсутствует четкая зависимость между концентрацией аммиака в крови и степенью ПЭ; при этом гипераммониемия выявляется у большинства больных циррозом печени (табл. 3) [13].

Лечение печеночной энцефалопатии
ПЭ – грозный признак поражения печени, имеющий важное прогностическое значение, поэтому необходимым является максимально раннее и адекватное лечение ПЭ [14]. Комплексное лечение ПЭ необходимо осуществлять в совокупности с этиотропной терапией (противовирусная терапия гепатотропных вирусных инфекций), патогенетическим лечением (медьэлиминирующая терапия при болезни Вильсона–Коновалова, иммунодепрессивная терапия при аутоиммунном гепатите, применение урсодезоксихолевой кислоты при холестатических заболеваниях печени и др.), а также симптоматической терапией (мочегонные препараты при отечно-асцитическом синдроме, коррекция других проявлений печеночноклеточной недостаточности) [15].
Общей рекомендацией по лечению ПЭ является необходимость соблюдения больными строгого постельного режима, что позволяет избегать вклада работы скелетной мускулатуры в гипераммониемию. Комплексное лечение ПЭ подразумевает воздействие на различные звенья патогенеза ее развития, в первую очередь направлено на снижение степени гипераммониемии [1, 11, 12].
1. Выявление и устранение факторов, способствующих развитию ПЭ (см. ниже).
2. Уменьшение степени гипераммониемии.
А. Уменьшение количества аммиакогенного субстрата:
• очищение желудочно-кишечного тракта (сифонные клизмы);
• уменьшение потребления белка.
Б. Связывание аммиака в крови:
• L-орнитин-L-аспартат;
• L-аргинин-L-яблочная кислота.
В. Подавление образования аммиака:
• нерезорбируемые антибиотики или по показаниям антибиотики широкого спектра действия;
• ацидификация кишечного содержимого лактулозой, ацидифицирующие клизмы.
Г. Постельный режим.
3. Восстановление баланса аминокислот (инфузии растворов аминокислот с разветвленной цепью).
4. Комплексная поддерживающая терапия (лечение инфекций, коррекция анемии и гипоксии, коррекция нарушений электролитного баланса и рН и др.).
Необходим тщательный поиск факторов, провоцирующих развитие ПЭ, поскольку нередко их устранение в короткие сроки позволяет ликвидировать симптомы ПЭ [16]. Это особенно важно в случае желудочно-кишечного кровотечения.
Факторы, провоцирующие развитие ПЭ у больных циррозом печени:
• желудочно-кишечное кровотечение из варикозно расширенных вен пищевода или язвенных дефектов слизистой оболочки желудочно-кишечного тракта;
• назначение психотропных препаратов;
• назначение препаратов, способных вызывать лекарственное поражение печени;
• форсированная диуретическая терапия (в первую очередь петлевыми диуретиками);
• употребление алкоголя;
• инфекции (в первую очередь спонтанный бактериальный перитонит, бронхо-легочные инфекции, инфекции мочевыводящих путей);
• наложение портокавальных анастомозов;
• избыточное употребление пищевого белка;
• хирургические вмешательства по поводу интеркуррентных заболеваний;
• лапароцентез с удалением большого количества асцитической жидкости (особенно без восполнения объема циркулирующей крови раствором альбумина);
• запоры.
Поскольку источником образования аммиака, не обезвреживающегося в печени и/или проникающего в системный кровоток через портокавальные шунты, является просвет желудочно-кишечного тракта, до настоящего времени самым эффективным методом уменьшения количества аммиакогенного субстрата остается очищение кишечника [17]. Этого достигают при использовании слабительных средств для очищения верхних отделов кишечника и клизм для нижних отделов. Пероральные слабительные лучше применять через назогастральный зонд – 10 %-ный раствор маннитола в объеме 500–1000 мл за 60–120 минут, 20 %-ный магния сульфат 50–100 мл. Однако эти препараты могут вызывать дегидратацию, гипонатриемию, гипокалиемию и метаболический алкалоз, что в свою очередь может способствовать развитию печеночной комы, поэтому в клинической практике применяются редко. Более доступно и наиболее адекватно удаление содержимого толстого кишечника с помощью сифонных клизм, что быстро уменьшает количество азотистого субстрата. Желательно использовать подкисленную воду (например, с добавлением 0,25–1,0 % уксусной кислоты) с целью связывания как можно большего количества аммиака. Нельзя применять мыльные и другие щелочные клизмы, поскольку они вызывают переход аммиака из просвета кишечника в кровеносное русло, где рН ниже. Наиболее целесообразно применение клизм с лактулозой: 300 мл лактулозы + 700 мл воды, а после них – с чистой водой.
Ограничение потребления белка с пищей – также доступный путь уменьшения образования аммиака в толстой кишке и снижение тем самым степени гипераммониемии [18]. При тяжелой степени ПЭ суточное потребление белка уменьшают до 20–30 г. В первые дни допустимо ограничение суточного количества потребляемого белка до 10 г, однако при более длительном ограничении возможен гиперкатаболизм эндогенных белков с нарастанием уровня аммиака в сыворотке крови. Необходимые для жизнедеятельности 1200 (до 2000) ккал восполняют приемом 5 %-ного раствора глюкозы внутрь или введением 10 %-ного раствора глюкозы внутривенно. После улучшения состояния больных содержание белка в диете можно постепенно увеличивать до 1 г/кг массы тела в сутки, обеспечивая его прирост на 10 г каждые сутки. Растительный белок переносится легче, чем животный; содержит небольшое количество метионина и ароматических аминокислот (при его приеме образуется меньше аммиака). Используют также молочный белок, содержащий в большом количестве аминокислоты с разветвленной цепью.
Другим путем уменьшения образования аммиака является подавление бактерий, способствующих образованию аммиака в просвете кишечника [19, 20]. Для выполнения этой задачи в терапии ПЭ могут быть использованы любые антибиотики широкого спектра действия, подавляющие кишечную флору, выделяющиеся с желчью или накапливающиеся в просвете кишечника в необходимой концентрации. С этой целью широко применяются фторхинолоны. Однако по возможности, особенно при начальных степенях ПЭ, необходимо проведение селективной деконтаминации кишечника путем назначения невсасываемых антибиотиков с широким спектром действия (на 4–7 дней, а иногда длительно – до 6–12 месяцев), что позволяет резко снижать уровень образования аммиака кишечными бактериями за счет санации кишечника. С этой целью применяется неомицин 4–6 г/сут или, более предпочтительно, рифаксимин 800–1200 мг/сут.
Слизистая оболочка кишечника человека не содержит ферменты, расщепляющие синтетические дисахариды, к которым относится лактулоза [21]. Перорально принимаемая лактулоза достигает слепой кишки, в которой она расщепляется бактериями с образованием преимущественно молочной кислоты. В результате рН каловых масс понижается. Это способствует росту бактерий, расщепляющих лактозу. При этом рост аммониегенных микроорганизмов, в частности бактероидов, подавляется. Лактулоза может “детоксицировать” жирные кислоты с короткой цепью, образующиеся при наличии крови и белков в просвете желудочно-кишечного тракта. В присутствии лактулозы и крови бактерии толстой кишки в основном расщепляют лактулозу. Это имеет особое значение при ПЭ, вызванной кровотечением. Кислая реакция каловых масс может уменьшать ионизацию и, следовательно, абсорбцию аммиака, а также аминов и других токсичных азотсодержащих соединений. В толстой кишке лактулоза более чем в 2 раза увеличивает образование растворимых соединений азота. В результате азот не абсорбируется в виде аммиака и уменьшается образование мочевины. При назначении лактулозы нужно стремиться к образованию кислого кала без диареи у больного. Препарат назначают в дозе 15–30 мл 2–3 раза в сутки, что приводит к 2- или 3-кратной дефекации (но не более) полужидким калом. К нежелательным явлениям относят метеоризм, диарею и боли в кишечнике.
Препараты, направленные на снижение концентрации аммиака в крови, улучшают обезвреживание аммиака в печени и связывают аммиак в крови [21, 22]. Ведущим гипоаммониемическим средством является L-орнитин-L-аспартат (Гепа-мерц). Гипоаммониемическое действие L-орнитина-L-аспартата связано с несколькими механизмами: орнитин стимулирует в перипортальных гепатоцитах карбамоил-фосфат-синтетазу I – ведущий фермент синтеза мочевины; аспартат и α-кетоглутарат стимулируют в перивенозных гепатоцитах, мышцах и головном мозге глутаминсинтетазу; орнитин и аспартат сами являются субстратами цикла синтеза мочевины (орнитин включается в цикл мочевины на этапе синтеза цитруллина, аспартат – на этапе синтеза аргининосукцината). Кроме этого в качестве положительного эффекта этих препаратов при ПЭ для больных циррозом печени рассматривается ингибирование катаболизма белка в мышцах, нормализация соотношения аминокислот в крови и антиоксидантный эффект. Эффективность L-орнитина-L-аспартата колеблется от 40 % при тяжелой ПЭ до 70–100 % при начальных признаках ПЭ.
Наиболее эффективна следующая схема лечения клинически выраженной ПЭ: внутривенные вливания по 20–40 г в сутки в течение 1–2 недель, затем пероральный прием препарата в дозе 9–18 г в сутки (по 3–6 г 3 раза в сутки после еды) в течение 2–3 недель. Больным ПЭ на фоне цирроза печени показано длительное (зачастую пожизненное) лечение этим препаратом.
Снижение уровня аммиака в крови достигается также парентеральным введением L-аргинина-L-яблочной кислоты (500 мл в сутки), участвующей в цикле трикарбоновых кислот и синтезе мочевины [23].
Развитие ПЭ сопровождается изменением соотношения между аминокислотами с разветвленной цепью и ароматическими аминокислотами [6]. Для лечения острой и хронической ПЭ применяются инфузии растворов, содержащих большую концентрацию аминокислот с разветвленной цепью. Механизм их действия связан с нормализацией аминокислотного состава крови. Наиболее широко эти препараты применяются больными циррозом печени с отсутствием белковой толерантности [24].
Важно подчеркнуть, что комплексная терапия ПЭ в сочетании с другими видами лечения (этиотропное, патогенетическое, симптоматическое) позволяет улучшать качество жизни больных циррозом печени различной этиологии и изменять их прогноз [25–27].



{kind=link}