
Введение
Истинная полицитемия (ИП, первичная полицитемия, эритремия) – хроническое прогрессирующее клональное миелопролиферативное заболевание с относительно благоприятным течением. ИП характеризуется гиперпролиферацией эритроидного, миелоидного и мегакариоцитарного ростков кроветворения с появлением спленомегалии в ранней (эритремической) фазе заболевания, развитием анемии и миелофиброза в фазе истощения кроветворения. Эритремия – редкое (орфанное) заболевание, которое развивается в основном у пациентов старшей возрастной группы [1]. Патогенез ИП связан с клональной пролиферацией неопластически трансформированной стволовой клетки: мутация в гене тирозинкиназы JAK2 (JAK2V617F) гранулоцитов [1, 2]. Актуальность патологии обусловлена риском развития тромботических и геморрагических осложнений, вероятностью трансформации в острый лейкоз [1, 3].
Для эритремии характерно наличие двух основных синдромов: плеторического и миелопролиферативного. Плеторический синдром обусловлен увеличением массы циркулирующих эритроцитов и проявляется головокружением, головными болями, ухудшением зрения, кожным зудом (особенно после приема ванны), стенокардией. Характерны сосудистые осложнения – тромбозы любой локализации, приступы покраснения пальцев рук и ног с болью и жжением (эритромелалгия). Миелопролиферативный синдром проявляется кожным зудом, потливостью, слабостью, повышением температуры тела, болями в костях. Повышенный распад гранулоцитов сопровождается нарушением пуринового обмена, что приводит к развитию гиперурикемии, мочекаменной болезни, подагрического артрита. При ИП в патологический процесс вовлекаются преимущественно костный мозг, селезенка и печень [1]. Сведения о патологии почек при ИП немногочисленные. В доступной литературе найдено описание всего 24 случаев развития нефропатии при данном заболевании. Как правило, проявления нефропатии включали протеинурию, микрогематурию, снижение клубочковой фильтрации. По данным нефробиопсии, выполненной 22 пациентам, наиболее часто встречались фокально-сегментарный гломерулосклероз и IgA-нефропатия. В двух случаях был диагностирован мембранопролиферативный гломерулонефрит, в одном – быстропрогрессирующий гломерулонефрит [4]. Клинико-лабораторная симптоматика включала протеинурию/нефротический синдром, микрогематурию. Нарушение почечной функции отмечено более чем у половины всех пациентов. За период наблюдения прогрессирование почечной недостаточности до терминальной стадии и потребность в диализе наблюдались у половины всех пациентов.
Патогенез формирования нефропатии при ИП может включать несколько механизмов. Во-первых, характерное для ИП увеличение объема и вязкости крови вызывает пассивное расширение капилляров и повреждение интимы, что влечет за собой образование микротромбов с окклюзией клубочковых капилляров, ишемию тканей и снижение клубочковой фильтрации. В отсутствие лечения ишемия сохраняется и, вероятно, приводит к хроническому повреждению почек. Кроме того, ИП часто ассоциируется с артериальной гипертензией и гиперурикемией, которые также влияют на почечную микроциркуляцию. Во-вторых, тромбоцитоз и аномальная активация мегакариоцитов могут быть критическим фактором развития гломерулярного склероза. Важную роль в этом процессе играют гиперпродукция тромбоцитарного фактора роста (PDGF) и трансформирующего фактора роста β (TGF-β) [4]. Пролонгированное высвобождение PDGF и других цитокинов индуцирует экстракапиллярную пролиферацию, а также стимулирует пролиферацию клубочковых мезангиальных клеток и образование внеклеточного матрикса. TGF-β индуцирует синтез коллагена и фибриногена через мезангиальные клетки, что ведет к мезангиальному склерозу и апоптозу подоцитов [4–7]. Характерная для ИП гиперурикемия ассоциируется с тубулоинтерстициальным поражением почек [1]. Полиморфизм клинических проявлений при ИП, включая почечную патологию, может стать причиной значительных трудностей при их трактовке, поэтому клинические наблюдения, иллюстрирующие поражение почек при истинной полицитемии, представляют безусловный интерес.
Клиническое наблюдение
Пациент Л. 59 лет госпитализирован в ревматологическое отделение в октябре 2019 г. с жалобами на слабость в ногах, сонливость, шаткость походки, головокружение, кожный зуд после водных процедур, наличие отеков ног, геморрагических высыпаний на коже нижних конечностей, местами с образованием язвочек, жжение в стопах, голенях до уровня нижней трети голеней. В анамнезе – повышение артериального давления (АД) до 150 и 90 мм рт.ст., базисная антигипертензивная терапия не проводилась. В феврале 2018 г. отметил утяжеление артериальной гипертензии с максимальным подъемом АД до 170 и 100 мм рт.ст.; выявлена протеинурия более 3,0 г/сут. При обследовании у нефролога в марте того же года протеинурия – 1,2 г/сут., гемоглобин крови – 142 г/л, эритроциты – 6,3×1012/л, тромбоциты – 402×109/л, лейкоциты – 12,8×109/л, уровень креатинина крови повышен до 287 мкмоль/л. Выявленные клинико-лабораторные симптомы первоначально трактовались в рамках гипертонической нефропатии, в дальнейшем – хронического гломерулонефрита. Назначены β-адреноблокаторы и препараты центрального действия. Через месяц наблюдения сохранялась протеинурия 1,0 г/сут., уровень креатинина снизился до 135 мкмоль/л. В октябре 2018 г. – визит-контроль: гемоглобин крови – 149 г/л, тромбоциты – 583×109/л, лейкоциты – 27,5×109/л, палочкоядерные нейтрофилы – 17%, креатинин крови – 140 мкмоль/л, мочевая кислота – 509 мкмоль/л. Рекомендована консультация гематолога. В то же время в связи с развитием артрита I плюснефалангового сустава ревматологом установлен диагноз «подагра, подагрический артрит», назначен преднизолон в дозе 30 мг/сут. коротким курсом с эффектом.
В феврале 2019 г. консультирован в клинике гематологии, с учетом данных генетического исследования (JAK2V617F в 14-м экзоне) установлен диагноз хронического миелопролиферативного заболевания, JAK2 положительного (истинная полицитемия). Назначена патогенетическая цитостатическая терапия (гидроксикарбамид – 500 мг/сут.), антиагреганты.
В марте 2019 г. возникла повторная атака острого артрита I плюснефалангового сустава. Вновь по рекомендации ревматолога принимал преднизолон 30 мг/сут., явления артрита были купированы. Осенью 2019 г. отметил появление обильных геморрагических высыпаний на коже голеней, склонных к слиянию, с образованием пузырей до 1 см, боли и припухания голеностопных суставов. Повторно консультирован ревматологом, диагноз «геморрагический васкулит, кожно-суставная, почечная форма». В связи с усилением болей в голеностопных суставах, слабостью в ногах, сохранением высыпаний с изъязвлениями на голенях был госпитализирован в ревматологическое отделение.
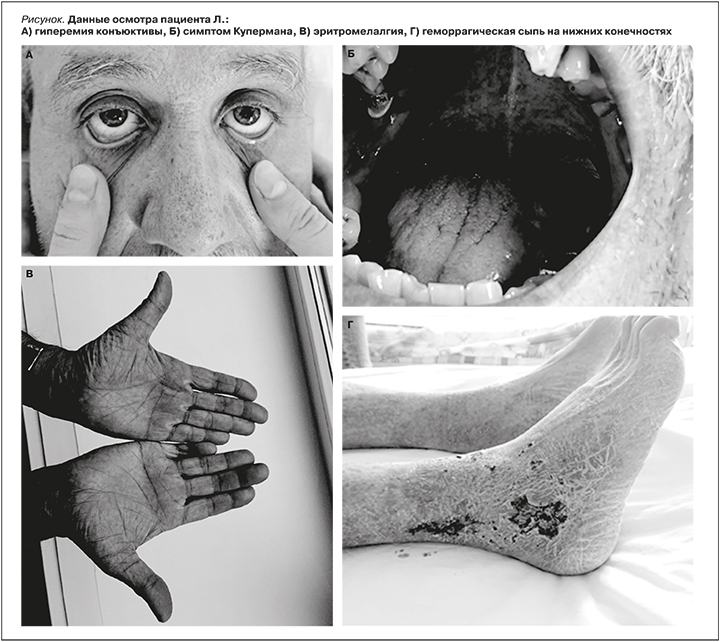
При объективном осмотре отмечены симптомы, характерные для ИП: положительный симптом Купермана, гиперемия конъюнктивы, эритромелалгия, геморрагические высыпания на голенях (см. рисунок). В периферической крови: эритроциты – 6,8×1012/л, гемоглобин – 180 г/л, лейкоциты – 26,69×109/л, тромбоциты – 410×109/л; повышение уровней креатинина крови до 199 мкмоль/л, мочевой кислоты до 621 мкмоль/л, гиперкоагуляция по данным коагулограммы. В анализах мочи мочевой осадок без патологии, суточная протеинурия – 0,12–0,6 г.
Маркеры аутоиммунных заболеваний отрицательные.
Проведен консилиум в составе врачей-ревматологов и нефрологов. Решение консилиума: «С учетом хронологически одновременного появления изменений в анализах мочи, полицитемии, нарушения функции почек имеется несомненная патогенетическая связь между заболеванием крови и поражением почек. Нарушение функции почек обусловлено, вероятнее всего, гиперурикемией в рамках гематологического заболевания. Верификация нефропатии возможна только при морфологическом исследовании почечного биоптата. Однако из-за опасности геморрагических осложнений, свойственных полицитемии, от проведения нефробиопсии воздержаться. Убедительных данных за васкулит как системного заболевания у пациента нет». Сформулирован окончательный диагноз «основное заболевание – хроническое миелопролиферативное заболевание (истинная полицитемия), мутация гена JAK2 V6617F в 14-м экзоне. Состояние на фоне терапии гидроксикарбамидом с февраля 2019 г. Вторичная подагра. Подагрический артрит, межприступный период. Вторичная нефропатия (гломерулонефрит? Интерстициальный нефрит?). Хроническая болезнь почек, стадия С3б. Вторичная артериальная гипертензия. Осложнения – хроническая почечная недостаточность 1-й стадии. Трофические язвы голеней».
В настоящий момент у пациента регистрируется частичный ответ на проводимую циторедуктивную терапию ИП. Клинически состояние пациента с положительной динамикой (геморрагические высыпания не рецидивировали, кожный зуд купирован, АД в нормотензивном диапазоне, азотемия сохраняется на прежнем уровне (креатинин крови – 181–157 мкмоль/л), мочевая кислота максимально до 375 мкмоль/л. Продолжается миелосупрессивная терапия гидроксикарбамидом, с целью профилактики тромботических осложнений применяются антитромбоцитарные препараты в сочетании с эпизодическими кровопусканиями, для контроля гиперурикемии используется аллопуринол.
Обсуждение
Особенности данного клинического наблюдения: развитие симптомов поражения почек в дебюте гематологического заболевания и присоединение в дальнейшем клинической симптоматики, свойственной ревматическим заболеваниям, в частности геморрагическому васкулиту. Трудности диагностики обусловлены сочетанием имевшейся почечной патологии, которая исходно трактовалась как гломерулонефрит, и появлением кожных геморрагических высыпаний. Наличие у пациента верифицированного гемобластоза не было должным образом оценено. Причины острого подагрического артрита как проявления возможной вторичной подагры ревматологом первично также не были рассмотрены. Использование полидисциплинарного подхода позволило оценить многообразие клинической симптоматики в рамках единого патологического процесса.
Заключение
Трудности диагностики нефропатии в рамках гематологического заболевания в представленном наблюдении обусловлены в т.ч. его редкой встречаемостью. Успешная диагностика редких болезней возможна при наличии у врача знаний, выходящих за пределы собственной специальности, а также при использовании специализированных знаний в рамках полидисциплинарного взаимодействия.


