
Боковой амиотрофический склероз (БАС) – неуклонно прогрессирующее нейродегенеративное заболевание, приводящее к неизбежной гибели пациента. Наиболее часто БАС дебютирует на шестом десятилетии жизни [1], в большом проценте случаев заболевание развивается и в более раннем возрасте. БАС – достаточно редкое заболевание. По данным Московского регистра, проведенного в 2006–2007 гг., частота БАС составила 1,16 случая на 100 тыс. населения [2]. БАС не единственное тяжелое, смертельное заболевание среди неврологических заболеваний, однако по тяжести его можно, пожалуй, поставить на первое место. Причина в сочетании нескольких моментов. Во-первых, на данный момент заболевание неизлечимо, по мере прогрессирования происходит потеря основных функций организма вплоть до тетраплегии, анартрии, афагии и неизбежно развивается дыхательная недостаточность с последующей остановкой дыхания и летальным исходом. Во-вторых, как правило, у пациентов с БАС практически не страдают критика и мышление. Сохранность интеллекта и осознание неизбежности летального исхода психологически усугубляют тяжесть заболевания.
Этиология БАС до настоящего времени полностью неизвестна. В 10% случаев заболевание имеет семейный характер, выявляются также спорадические мутации. Генов, ассоциированных с развитием БАС, к настоящему моменту выявлено более 20. Клиническая картина включает обязательное поражение верхнего и нижнего мотонейронов, выявляются различия по локализации и возрасту дебюта, скорости прогрессирования, представленности экстрамоторной симптоматики. Подобная вариативность затрудняет диагностику заболевания и позволяет задуматься о различных этиопатогенетических факторах развития заболевания. Выявлены особенности течения БАС у пациентов с различными мутациями. Например, мутация в гене, кодирующем эндосомальный фактор обмена гуанина (GEF), вызывает медленнопрогрессирующую форму заболевания с ранним дебютом. Мутация в гене, кодирующем ДНК/РНК-связывающий белок, FUS-мутация, характеризуется очень быстрым прогрессированием, бульбарным или диффузным дебютом [4]. На данный момент наиболее часто у пациентов с семейным и спорадическим БАС встречается обнаруженная в 2011 г. мутация в гене C90RF72 – экспансия GGGGCC [5]. По литературным данным, у пациентов с данной мутацией обнаруживается преимущественно быстрый темп прогрессирования БАС, бульбарный дебют, в 50% случаев при этой мутации выявляются когнитивные нарушения. Мутация в гене C90RF72 характерна также для фронто-темпоральной деменции (ФТД) [5].
На мутацию C90RF72 обследованы 48 пациентов с БАС, достоверным по Эль-Эскориальским критериям. Применялись следующие методы генетического обследования: метод фенольно-хлороформной экстракции ДНК из венозной крови, полимеразной цепной реакции (ПЦР), фрагментный анализ одноцепочечного конформационного полиморфизма (SSCP) ДНК, Саузерн блоттинг. Мутация в гене C90RF72 выявлена у 2 (4%) пациентов. Ниже приведены клинические наблюдения этих пациентов, отражающие особенности течения БАС при наличии данной мутации.
Клинический случай 1
Пациент Г. 58 лет обратился с жалобами на постепенно прогрессирующее нарушение речи, глотания, повышенное слюноотделение, слабость в левых конечностях.
Заболевание дебютировало 11 месяцев назад с появления дисфонии, замедленности речи, затем присоединились нечеткость речи и через месяц пациент стал поперхиваться при глотании. Через несколько месяцев присоединились повышенная саливация, слабость левой ноги. Симптомы постепенно прогрессировали до грубой дисфагии, дизартрии, нарастала слабость в левой ноге – преимущественно в стопе, присоединилась слабость в левой кисти. Появилась плаксивость, эмоциональная лабильность.
Из анамнеза известно, что хронических заболеваний, профессиональных вредностей, острых и привычных интоксикаций пациент не имел, укусы клещей отрицал. До настоящего заболевания считал себя практически здоровым.
Наследственный анамнез: отцу пациента был поставлен диагноз БАС с бульбарным дебютом заболевания.
На момент первого обращения в клинику (длительность заболевания – 11 месяцев) в неврологическом статусе отмечены легкая дизартрия, замедленность речи, атрофия и фасцикуляции языка, дисфагия и дисфония, гнусавость и глухость голоса, затруднения проглатывания твердой пищи, потребность в изменении консистенции пищи. Правосторонний гемипарез со снижением мышечной силы до 4 баллов в руке (преимущественно в кисти) и 3 баллов в ноге на фоне резко оживленных сухожильных и периостальных рефлексов с расширением рефлексогенных зон, значительного повышения мышечного тонуса по спастическому типу в правых конечностях. Чувствительных и координаторных нарушений отмечено не было. Умеренные генерализованные спонтанные фасцикуляции мышц туловища и конечностей. Также у пациента присутствовали признаки когнитивной дисфункции: снижение критики к своему состоянию, некоторая эйфоричность, общая замедленность интеллектуальной деятельности. Грубые мнестические нарушения отсутствовали.
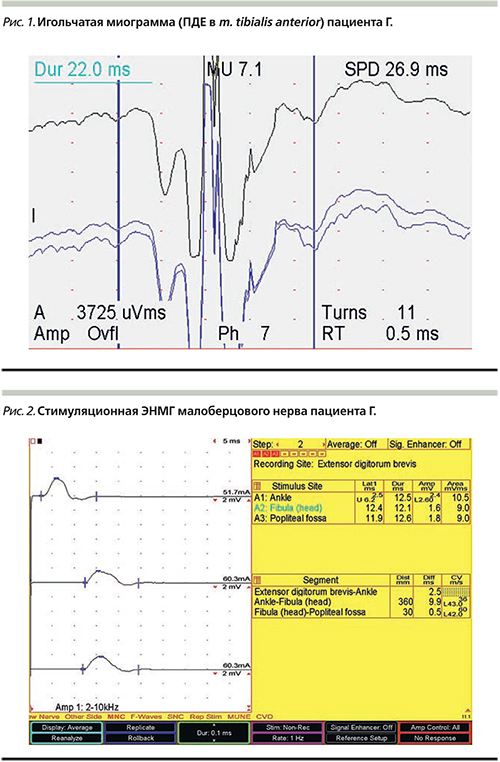
При первом обращении была выполнена игольчатая и стимуляционная электронейромиография (ЭНМГ): в мышцах бульбарного уровня, шейного и поясничного утолщений спинного мозга выявлена умеренная спонтанная активность в виде потенциалов фибрилляций (ПФ) и фасцикуляций (ПФЦ), положительных острых волн (ПОВ); увеличение средней амплитуды и длительности потенциалов двигательных единиц (ПДЕ) – текущая генерализованная нейронопатия (рис. 1). При стимуляционной ЭНМГ выявлено аксонально-демиелинизирующее поражение моторных волокон при сохранном сенсорном проведении (мотосенсорная диссоциация) (рис. 2).
При дальнейшем наблюдении отмечена отрицательная динамика в виде нарастания тетрапареза, выраженности дисфагии и дизартрии. Уровень когнитивных расстройств был неизменным. Через 7 месяцев после первичного обращения у пациента выявлено нарастание дисфагии – присоединились нарушения проглатывания жидкой пищи (принимал жидкую пищу только с загустителями), наросла дизартрия – речевой контакт с больным значительно затруднился, увеличился парез левой стопы до 2 баллов, присоединилась слабость проксимальной мускулатуры левой ноги. При дальнейшем прогрессировании заболевания углубилась выраженность дисфагии (однако пациент съедал необходимый объем пищи) и дизартрии (вплоть до анартрии), прогрессировали двигательные расстройства до тетраплегии. Через 33 месяца от дебюта заболевания наступил летальный исход на фоне нарастающей дыхательной недостаточности.
При генетическом обследовании у пациента была выявлена мутация в гене C90RF72. Несмотря на наследственный характер заболевания (наличие БАС у отца пациента), родственники пациента отказались от генетического исследования.
Таким образом, у данного пациента выявлен семейный быстропрогрессирующий БАС с бульбарным дебютом, когнитивной дисфункцией, ранним присоединением дыхательных нарушений, ассоциированный с мутацией C90RF72.
Клинический случай 2
Пациент С. 43 лет обратился с жалобами на слабость в руках и подергивание в мышцах конечностей и туловища.
Заболевание дебютировало 16 месяцев назад с появления фасцикуляций в правой руке, затем они появились с другой стороны. Через 4 месяца обратился за медицинской помощью к неврологу в поликлинику по месту жительства, был заподозрен БАС и пациенту было назначено обследование – ЭНМГ, магнитно-резонансная томография (МРТ) головного мозга и шейного отдела позвоночника. Пациент выполнил обследование, был направлен на кафедру для подтверждения диагноза БАС.
Из анамнеза известно, что до настоящего заболевания хронических заболеваний, профессиональных вредностей, острых и привычных интоксикаций не имел. Укусы клещей отрицает. Семейный анамнез БАС отрицает.
В неврологическом статусе при первом обращении отмечены насильственная неконтролируемая эмоциональность, легкая слабость мимической мускулатуры без бульбарных или речевых нарушений, атрофий и фасцикуляций лица и языка не выявлено. Верхний парапарез со снижением мышечной силы до 3 баллов с выраженными атрофиями верхнего плечевого пояса, оживлением глубоких рефлексов с расширением рефлексогенных зон. Мышечный тонус значимо не изменен. Чувствительных и координаторных нарушений не было. Когнитивных нарушений не выявлено на протяжении заболевания.
Результаты обследования: МРТ шейного отдела позвоночника – остеохондроз, МРТ головного мозга – ретроцеребеллярная арахноидальная киста, расширение периваскулярных пространств.
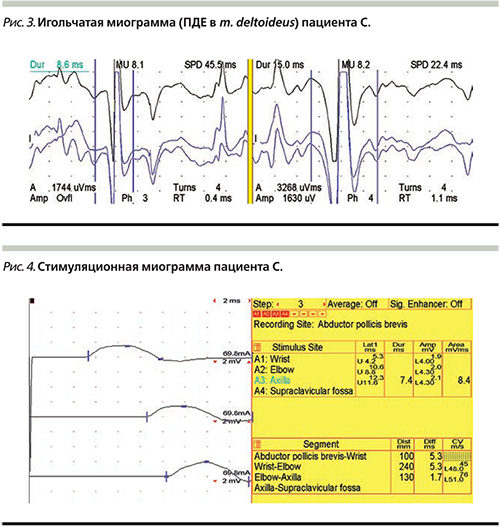
При игольчатой ЭМГ выявлена бурная спонтанная активность в виде ПФЦ, ПФ; увеличение средней длительности ПДЕ на 30%, увеличение амплитуды ПДЕ – текущая генерализованная нейропатия (рис. 3). При стимуляционной ЭНМГ выявлена аксонопатия моторных волокон при сохранности сенсорного проведения (мотосенсорная диссоциация) (рис. 4).
При дальнейшем наблюдении отмечено прогрессирование заболевания – нарастала слабость в верхних конечностях (практически до плегии), появилась слабость мышц туловища и дыхательной мускулатуры, нарастали дыхательные нарушения. Через 28 месяцев от дебюта заболевания наступил летальный исход на фоне нарастающей дыхательной недостаточности.
При генетическом исследовании выявлена мутация C90RF72. Генетическое исследование родственников пробанда (матери, сестры, дочери сестры, сына) выявило мутацию C90RF72 у сестры (41 год) и сына (17 лет) пациента. На момент исследования признаков неврологического поражения у родственников не наблюдалось. Отец пациента умер в возрасте до 50 лет.
Таким образом, у пациента выявлен семейный быстропрогрессирующий БАС с шейным дебютом, ранним присоединением дыхательных нарушений, ассоциированный с мутацией C90RF72.
Обсуждение
Мутация в гене С9ORF72 выявлена у 2 пациентов с подтвержденным семейным БАС (с семейным анамнезом заболевания и с выявленной мутацией у ближайших родственников), с различной проекцией дебюта (бульбарный и шейный), несколько отличающимися вариантами течения (значительная выраженность пирамидной симптоматики у пациента с бульбарным дебютом, классический вариант БАС с равномерным поражением верхнего и нижнего мотонейронов у пациента с шейным дебютом). Объединяет их выживаемость менее 3 лет (33 и 28 месяцев), быстрая генерализация процесса с присоединением слабости аксиальной мускулатуры и тяжелой дыхательной недостаточности. Также у пациентов выявлены экстрамоторные симптомы, часто обнаруживаемые у пациентов с данной мутацией: когнитивные нарушения у пациента Г. с бульбарным дебютом, эмоциональная лабильность у пациента С. с шейным дебютом.
В заключение можно сказать, что в последнее время очень активно ведутся исследования мутации C90RF72 у пациентов с БАС и ФТД во всем мире, что связано с наибольшей встречаемостью этой мутации среди всех ассоциированных мутаций с семейным БАС и ФТД. С учетом разнообразия клинической картины БАС выявление клинических особенностей заболевания, характерных для той или иной мутации, позволит целенаправленно подойти к генетическому анализу и генетическому консультированию, приблизиться к пониманию генетико-фенотипичеких особенностей заболевания.


