
Введение
На протяжении многих лет существовало устоявшееся мнение, согласно которому коронавирусы, поражающие человека, вызывают нетяжелые респираторные инфекции, доля которых в популяции составляет от 15 до 30% всех случаев острой респираторной вирусной инфекции [1]. Считалось, что у молодых людей в большинстве случаев такие инфекции практически не требуют специфического вмешательства и разрешаются самостоятельно, в то время как лицам старшей возрастной группы и коморбидным пациентам могло потребоваться лечение в стационаре [2]. Тем не менее за последние два десятилетия мы стали свидетелями появления в человеческой популяции циркуляции коронавирусов, вызывающих тяжелые повреждения органов и систем и тем самым опасных для жизни. В 2002 г. выявлен SARS-CoV (Severe Acute Respiratory Syndrome-Related coronavirus, тяжелый острый респираторный синдром, более известный как атипичная пневмония). Спустя десятилетие в 2012 г. в Саудовской Аравии зарегистрирована вспышка MERS-CoV (Middle East Respiratory Syndrome-Related Coronavirus, ближневосточный респираторный синдром).
В 2019 г. выявлен SARS-СоV-2, вызывающий тяжелое заболевание у человека. Всемирная организация здравоохранения 11.02.2020 присвоила официальное название инфекции, вызванной новым коронавирусом, COVID-19 («Coronavirus disease 2019»), а уже 11.03.2020 объявила о статусе пандемии новой коронавирусной инфекции [3].
Летальность от COVID-19 относительно не высока и колеблется от 1 до 5%. Тем не менее наличие кардиальной патологии у лиц с COVID-19 является одним из наиболее значимых предикторов неблагоприятного прогноза, что заставило выделить в особую группу риска пациентов с сердечно-сосудистыми заболеваниями (ССЗ) [4]. Так, суммарно на долю сердечной патологии (сердечная недостаточность) приходится примерно 40% всех случаев смертей пациентов с COVID-19: ~53% – дыхательная недостаточность; ~33% – сочетание дыхательной и сердечной недостаточности; ~7% – сердечная недостаточность [3]. Следует отметить, что летальность при наличии ССЗ отмечена в большей степени у пациентов пожилого и старческого возраста, что ассоциировано с большей распространенностью ССЗ в данных возрастных группах, функциональными нарушениями иммунной системы, а также с более частыми явлениями кардиотоксичности на фоне сниженного метаболизма при проведении этиотропной терапии коронавирусной инфекции.
Особенности SARS-СоV-2
SARS-СоV-2, подобно SARS-СоV и MERS-CoV, вызывает респираторную инфекцию, которая часто приводит к вирусной пневмонии и острому респираторному дистресс-синдрому (ОРДС).
Филогенетические исследования показали, что все три высокопатогенных коронавируса (SARS-CoV, MERS-CoV и SARS-CoV-2) принадлежат к роду Betacoronavirus [5, 6, 8]. Два штамма вируса, SARS-СоV и SARS-СоV-2, похожи: филогенетический анализ показал 79,6% идентичной геномной последовательности [9, 6, 8]. С учетом этой гомологии предполагается, что SARS-CoV-2 имеет много общих биологических свойств с SARS-CoV, что позволяет применять накопленные знания о жизненном цикле и патогенезе SARS-CoV для понимания SARS-CoV-2 [10, 11]. Для проникновения в клетку-хозяина оба вируса используют свои спайк-белки. Единица S1 белка S прикрепляется и связывается с рецептором клетки-хозяина. Затем для входа в клетку требуется праймирование белка S-клеточными протеазами клетки-хозяина, что приводит к слиянию оболочки вируса и мембраны клетки-хозяина и проникновению в эндотелиоцит.
Вместе с тем S-белок SARS-CoV-2 содержит две отличительные особенности, которых нет у SARS-CoV. Во-первых, он имеет более высокое сродство к человеческому ангиотензинпревращающему ферменту-2 (АПФ-2) [9, 11] и может напрямую связываться с АПФ-2 человека с аналогичной или даже более высокой аффинностью, чем у SARS-CoV [12]. Это высокое сродство к связыванию, вероятно, объясняет более высокую контагиозность SARS-CoV-2 и тяжесть течения COVID-19. Во-вторых, S-белок SARS-CoV-2 имеет вставку из четырех аминокислотных остатков на границе между субъединицами S1 и S2, что вводит новый сайт расщепления фурина, отсутствующий у других коронавирусов. Хотя функция этого нового сайта расщепления до настоящего момента не выяснена, аналогичные участки расщепления описаны у высокопатогенных вирусов птичьего гриппа и вируса болезни Ньюкасла [9, 11]. Имеется гипотеза, согласно которой именно эта примечательная особенность играет значительную роль в расширении клеточного или тканевого тропизма SARS-CoV-2 [9], способствуя полиорганным эффектам COVID-19.
Рецептором клетки-хозяина является АПФ-2, а используемой сериновой протеазой – TMPRSS2 [11]. Внедрение вирусной РНК вызывает выход клеточной в кровоток АПФ-2 и нарушение функционирования рецепторов к АПФ-2. В норме АПФ-2 едва присутствует в кровообращении в растворимой форме, тем не менее он широко экспрессируется, а его рецепторы обнаружены в эндотелиоцитах артерий и вен, клетках гладкой мускулатуры артерий почти всех органов, включая легкие, сердце, почки и головной мозг. Наиболее вероятно, что именно благодаря столь обширному представительству рецепторов к АПФ-2 мы имеем картину множественного повреждения органов и систем, в частности сердца и сердечно-сосудистой системы (ССС).
Особенностью SARS-CoV-2 (в дополнение к респираторным проявлениям) является способность провоцировать «цитокиновый шторм», в результате которого провоспалительные цитокины и хемокины, такие как фактор некроза опухоли-α (ФНО-α), интерлейкин-1β (ИЛ-1β) и ИЛ-6, производятся иммунной системой в чрезмерном количестве, что приводит к полиорганному повреждению [13]. Кроме того, COVID-19 вызывает нарушения свертывания крови у значительной части пациентов, что служит предпосылкой к тромбоэмболическим событиям [13, 11].
Другая отличительная черта SARS-СоV-2 – способность значимо влиять на функционирование ССС. Клинические данные показывают, что наличие у пациентов хронических заболеваний сердца и сосудов способствует более тяжелому течению COVID-19 [15, 16, 17] и связано со значительным увеличением смертности [18, 19, 8]. Помимо ухудшения течения и прогрессирования хронических заболеваний COVID-19 способствует развитию ССЗ de novo: острое и хроническое повреждение миокарда, аритмии, острый коронарный синдром и тромбоэмболии, сердечная недостаточность [5, 20].
COVID-19 имеет различные сердечно-сосудистые проявления, включая изменение сердечных биомаркеров (из-за ишемических или неишемических причин), миокардит, аритмию, кардиогенный шок, остановку сердца и тромбоэмболию. Ишемическое повреждение может приводить к инфаркту миокарда и повреждению сердца с диссеминированным внутрисосудистым свертыванием крови. Неишемическое повреждение миокарда приводит к миокардиту, кардиомиопатии, вызванной стрессом, и повреждению миокарда с синдромом высвобождения цитокинов (рис. 1) [49].
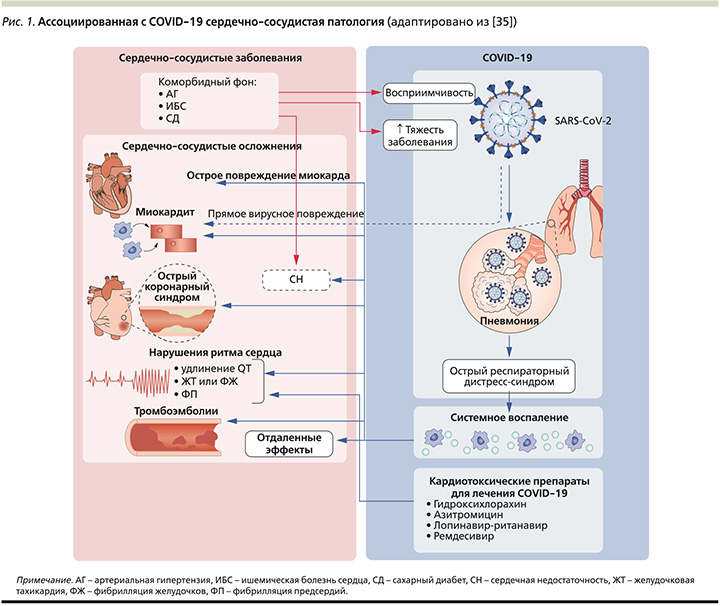
SARS-CoV-2 и ССС
Точные механизмы повреждения сердца и сосудов при COVID-19 установлены недостаточно полно. Отсутствуют четкие объяснения вариабельности и выраженности кардиологических проявлений. Неизвестно, почему у части больных ССС поражается, а у других нет.
Рассматриваются различные патофизиологические механизмы, приводящие к дисфункции: нарушение работы ренин-ангиотензин-альдостероновой системы (РААС), непосредственное кардиотоксическое действие вируса, поражение микроциркулярного русла, нарушение свертываемости крови, провоспалительное и цитокиновое воздействие на миокард (рис. 2), дыхательная дисфункция и гипоксия, кроме того, необходимо учитывать кардиотоксичность препаратов, применяемых при лечении COVID-19.
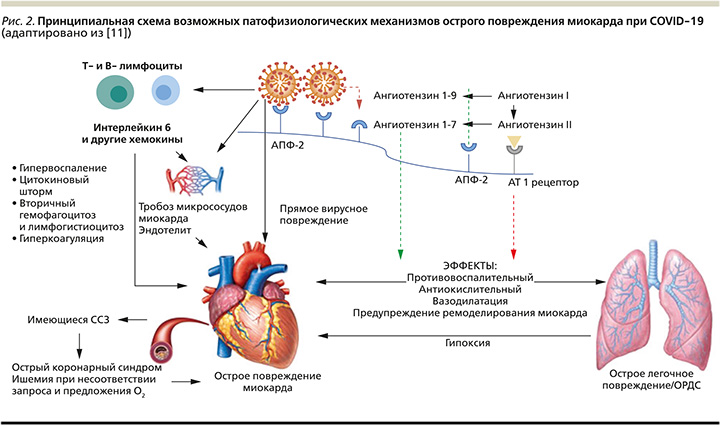
Дисфункция РААС
АПФ-2 – жизненно важное звено РААС, вовлекаемое в патогенез АГ и других ССЗ. Известно, что АПФ-2 представляет собой трансмембранный белок I типа, который экспрессируется в легких (высокий уровень экспрессии на поверхности альвеолярных клеток II типа), кардиомиоцитах, сердечных фибробластах и коронарных эндотелиальных клетках, эндотелии артерий и вен, гладкомышечных клетках артерий, в изобилии обнаружен в слизистой оболочке полости рта и носа, клетках энтероцитах малого таза, кишечника, почечных подоцитах и клетках проксимального извитого канальца [24].
Физиологическая роль АПФ-2 первично связана с расщеплением ангиотензина (АТ)-I до неактивного пептида АТ1-9, и с деградацией АТ-II в АТ1-7, связывающегося с Mas-рецепторами. АТ1-7 обеспечивает вазо- и кардиопротекцию, антипролиферативный, противовоспалительный и натрийуретический эффекты, а также обладает защитными свойствами в отношении сердечной недостаточности, тромбоза, гипертрофии миокарда, фиброза, аритмии, атерогенеза [24]. Расщепляя АТ-II, АПФ-2 ослабляет негативные эффекты последнего (вазоконстрикцию, цитокиноподобную активность, задержку натрия и развитие фиброза). Повышение уровня АТ-II по сравнению с уровнем АТ1-7 вызывает негативное влияние на сердце.
В каскад прямого вирусного повреждения сердца вовлечены рецепторы и сигнальные пути АПФ-2. При достаточной вирусной нагрузке экспрессия рецепторов АПФ-2 снижается, что приводит к дисрегуляции системы РААС [26, 4, 17] и вызывает негативные эффекты (вазоконстрикцию, цитокиноподобную активность, задержку натрия и развитие фиброза), а также содействует чрезмерной активации иммунной системы, поддерживающей состояние хронического воспаления в сосудистой стенке и тканях почек.
Непосредственное цитопатическое действие вируса на кардиомиоциты
По данным гистологического исследования выявлено, что у 35% пациентов с COVID-19 геном вируса обнаружен в тканях сердца. Кроме того, обнаружено заметное увеличение инфильтрации макрофагами с признаками повреждения кардиомиоцитов, что позволяет предположить, что вирус может непосредственно поражать ткани сердца [27].
С учетом высокой степени геномной гомологии между SARS-CoV, SARS-CoV-2 и схожего патогенеза обоснованной выглядит гипотеза, согласно которой SARS-CoV-2 может поражать миокард, напрямую проникая в кардиомиоциты, используя рецепторы к АПФ-2, представленные в достаточном количестве на их мембране. В пользу такого механизма свидетельствуют сообщения о миокардитах с фульмитантным течением при COVID-19, в частности, у пациентов без выраженных общевоспалительных и респираторных симптомов.
Необходимо отметить, что TMPRSS2 высоко экспрессируется в легких и почках, но в сердце и кровеносных сосудах присутствует в низких или умеренных уровнях, что указывает на участие и других механизмов в повреждении миокарда.
Провоспалительное и цитокиновое влияние
Согласно позиции American College of Cardiology/American Heart Association, главным механизмом острого поражения сердца при COVID-19 является патологический системный воспалительный ответ, проявляющийся «цитокиновым штормом».
«Цитокиновым штормом» названа крайняя степень иммунной дисрегуляции, выявляемой у некоторых больных COVID-19. Другое название этого расстройства – «синдром высвобождения цитокинов» (CRS). Возникает он при различных тяжелых соматических состояниях, включая бактериальный сепсис, а также у пациентов, получающих терапию Т-клеточными рецепторами химерного антигена (CAR), и гемофагоцитарный синдром [28]. CRS вызывает усиление системного воспаления, повышение проницаемости сосудов, что способствует образованию плевральных выпотов и отеков, а также приводит к внутрисосудистому истощению за счет потери жидкости в третьем пространстве и гипотензии. Совместно с вирус-индуцированным воспалением CRS создает дестабилизацию атеросклеротической бляшки, как следствие – возможен разрыв, приводящий к развитию острого инфаркта миокарда 1-го типа [3, 4]. Схожая ситуация наблюдается при тяжелых соматических состояниях, включая бактериальный сепсис, а также у пациентов, получающих терапию Т-клеточными рецепторами химерного антигена и при гемофагоцитарном синдроме [28].
Результаты ретроспективного многоцентрового исследования подтвердили, что воспалительные маркеры, включая повышенный ферритин и ИЛ-6, связаны с более тяжелым течением COVID-19, и указывают, что системное воспаление может быть существенным драйвером полиорганных повреждений [29, 19]. Другая группа исследователей также подтвердили, что у пациентов с тяжелым заболеванием имеется повышенный уровень сывороточных цитокинов ИЛ-2, -6, -10 и ФНО-α [30]. Системное высвобождение цитокинов, характеризующееся повышением уровней ИЛ-2, -6, -10, GCSF, интерферон-γ, MCP-1, MIP-1-α и ФНО-α, вероятно, способствует повреждению сердца в ситуации, аналогичной кардиотоксичности в условиях химерной антигенной рецепторной (CAR)-Т-клеточной терапии. Известно, что повреждение миокарда и систолическая дисфункция левого желудочка часто выявляются после применения CAR-T [31].
Точный механизм, посредством которого цитокины/хемокины повреждают миокард, неизвестен, но гибель кардиомиоцитов и эндотелиальных клеток в присутствии воспалительных цитокинов, таких как ФНО-α, хорошо документирована в литературе [32, 33].
Huang et al. обнаружили, что во время инфекции SARS-CoV-2 увеличение провоспалительных факторов, таких как ИЛ-1β, интерферон-γ, интерферон-индуцируемый белок-10 и моноцитарный хемоаттрактантный белок-1, может вызывать активацию хелперного Т-лимфоцита 1 (адаптивный Т-хелпер 1). По сравнению с больными легкой формой COVID-19 концентрации провоспалительных цитокинов в плазме тяжелых пациентов были значительно выше, что свидетельствует о взаимо-связи наличия «цитокинового шторма» и тяжести течения заболевания.
В том же исследовании отмечено недостаточное увеличение концентрации T-хелперов-2 и ИЛ-4, которые должны снижать степень выраженности воспалительного ответа. Это является реакцией организма на чрезмерное воспаление и указывает на роль SARS-CoV-2 в дисбалансе между клетками Th1 и Th2. Таким образом, «цитокиновый шторм» может быть одним из важных механизмов повреждения миокарда, а также важным механизмом полиорганной недостаточности, вызванной иммунным дисбалансом [54].
Другой предполагаемый провоспалительный механизм COVID-19 связан со взаимопотенцирующим иммунным ответом на дисбаланс РААС [1, 36]. Повышенные концентрации АТ-II вызывают негативные эффекты (вазоконстрикцию, цитокиноподобную активность, задержку натрия и развитие фиброза) и содействуют чрезмерной активации иммунной системы, поддерживающей состояние хронического воспаления. В экспериментальных, проспективных и интервенционных исследованиях выявлено взаимодействие некоторых цитокинов с системами, регулирующими артериальное давление (РААС, симпатической нервной системой) и развитием АГ. Например, ИЛ-6, рассматриваемый в качестве предиктора неблагоприятных исходов при COVID-19, – один из ключевых цитокинов в инициации иммуновоспалительного ответа при АГ [37]. В другом исследовании на моделях животных определена связь АГ с циркулирующими CD8+ T-клетками, в то время как в условиях инфицирования SARS-CoV-2 чрезмерно высокий уровень циркулирующих CD8+-цитотоксичных T-клеток содействует гиперпродукции провоспалительных цитокинов [37].
Дыхательная дисфункция и гипоксия
Необходимо помнить, что коронавирусы – это прежде всего семейство вирусов, поражающих дыхательные пути. Слабопатогенные вирусы HCoV (включающие HCoV-229E, HCoV-OC43, HCoV-NL63 и HCoVHKU) имеют тропность к верхним отделам дыхательных путей, вызывая легкие и умеренные сезонные респираторные заболевания, а высокопатогенные коронавирусы (SARS-CoV, MERS-CoV и SARS-CoV-2) поражают нижние отделы [37, 11].
На основании данных современной литературы предполагается, что клеточное внедрение и последующее распространение вируса в дыхательную систему приводит к следующим процессам: 1) увеличению соотношения уровней АПФ-1 и АТ-II по сравнению с уровнем АПФ-2 и АТ1-7; 2) значительному воспалительному ответу, опосредованному нейтрофилами, макрофагами и CD8+-Т-клетками, ведущему к альвеолярному отеку; 3) образованию тромбов; 4) потенциальному разрушению пневмоцитов II типа [24, 29, 38, 39, 40]. Все эти процессы зачастую приводят к формированию острого повреждения легких, острого респираторного дистресс-синдрома, CRS, ДВС-синдрома и как следствие – к тяжелой дыхательной недостаточности [1].
Другой потенциальный механизм гипоксии был предложен W. Liu et al. [41]. Обнаружено, что при COVID-19 синтезируются белки ORF8, ORF10, orf1ab, ORF3a и поверхностные гликопротеины, которые взаимодействуют с гемовым компонентом гемоглобина. Белки ORF8 обладают способностью связываться с порфирином в геме. Белки ORF10, orf1ab и ORF3a могут отделять железо от молекулы гема цепи гемоглобина-β1 (рис. 3). Таким образом, гемоглобин теряет способность эффективно доставлять кислород [41] и формирует гемическую гипоксию [26, 41]. Инактивация гемоглобина приводит к тому, что эритроциты теряют реологию и деформируются. Это потенциально является еще одним фактором, способствующим гипоксии и тромбозу в микроциркуляции.
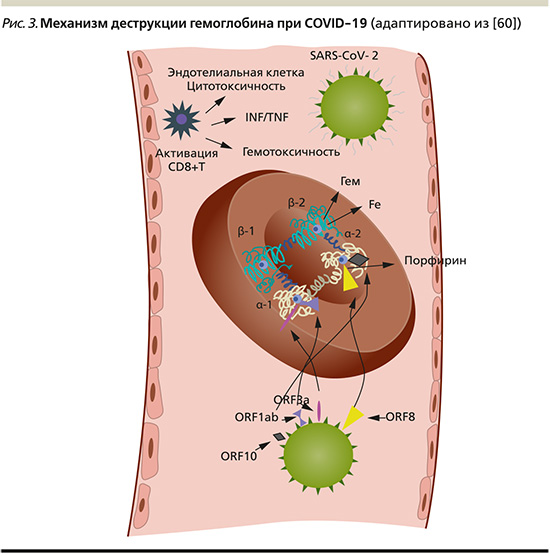
На фоне дыхательной, тканевой и гемической гипоксии возникает дисбаланс между возросшими метаболическими потребностями миокарда и снижением сердечного резерва, что приводит к дисфункции, а затем и к повреждению кардиомиоцитов.
Эндотелиальная дисфункция
К особенностям SARS-CoV-2 относятся индукция выраженной воспалительной реакции и более выраженное поражение эндотелиальных клеток, определяемое на основании анализа экспрессии ИЛ-6, ФНО-α, внутриклеточной молекулы адгезии-1 и каспазы-1 [13, 43].
Причиной эндотелиальной дисфункции (ЭД) у больных COVID-19 могут быть как CRS, так и иммунно-опосредованное поражение эндотелиоцитов.
Цитокины и белковые провоспалительные медиаторы служат ключевым фактором, способствующим нарушению эндотелиальной функции [44]. Так, при протеомном анализе 185 маркеров, отражающих воспаление и дисфункцию эндотелия в системном кровотоке, показано, что наличие «цитокинового шторма» сочеталось с диффузным поражением сосудистого эндотелия [44]. Повышение содержания провоспалительных цитокинов у больных COVID-19 прямо коррелирует с увеличением уровней маркеров, отражающих системное сосудистое поражение по типу васкулита, и маркеров ремоделирования сосудистого русла [44]. Кроме этого имеется связь между клинической тяжестью заболевания со степенью выраженности ЭД [36]. При COVID-19 отмечается сдвиг гемостаза в прокоагулянтную сторону, что находит отражение в повышении уровня фибриногена, продуктов распада фибрина, D-димера и фактора фон Виллебранда, и это коррелирует с тяжестью заболевания и риском тромбозов, что является отражением ЭД [48].
Генерализованная ЭД, возникающая под влиянием SARS-CoV-2, приводит к массивному тромбообразованию и закупорке мелких сосудов легких, почек, сердца, печени и других органов микротромбами, что вызывает нарушение микроциркуляции в данных органах и, соответственно, нарушение их функции. Микротромбоз легочных микрососудов приводит к нарушению перфузии легких и усугублению дыхательной недостаточности.
Дополнительную тяжесть состоянию могут добавлять вирусиндуцированные аутоиммунные реакции. Известно, что ИЛ-6 среди других интерлейкинов вызывает гиперактивность тромбоцитов, их слипание и агрегацию [49]. Кроме того, опосредованная ФНО экспрессия тканевого фактора способствует агрегации тромбоцитов и образованию мультифокальных тромбов и межальвеолярной коагуляции. Это происходит потому, что ФНО активирует сосудистые моноциты и сосудистые эндотелиальные клетки для экспрессии тканевого фактора на их клеточной поверхности. Таким образом активируется внешний путь системы свертывания крови, вызывая образование микротромба. Другим путем, посредством которого ФНО способствует образованию микротромба, считается снижение эндотелиальной экспрессии тромбомодулина и гликозаминогликанов, регулирующих систему свертывания крови [50, 49].
Все эти нарушения регуляции могут приводить к диссеминированному внутрисосудистому свертыванию крови (ДВС-синдрому). ФНО и другие цитокины активируют нейтрофилы для высвобождения различных медиаторов воспаления, таких как внеклеточная ловушка нейтрофильных клеток, гистоны, лейкотриен В4, металлопротеиназа матрикса, миелопероксидаза, активные формы кислорода и другие, способные дополнительно повреждать эндотелиальные клетки [50].
ДВС-синдром сопровождается образованием фибриновых тромбов как в микроциркуляторном русле, так и в более крупных сосудах – венах нижних конечностей, что приводит к возникновению тромбоэмболии легочной артерии, первичному тромбозу легочных и коронарных артерий, артерий и вен головного мозга и кишечника, правых отделов сердца. В целом проявления ДВС-синдрома при COVID-19 носят классический характер с развитием не только тромбозов, но и геморрагий в виде кровоизлияний в тканях и диапедеза эритроцитов
Современное понимание взаимопотенцирующего воздействия звеньев патогенеза
Однозначно выделить ведущий патогенетический механизм негативного влияния SARS-СoV-2 на организм на текущий момент не представляется возможным ввиду одномоментного по времени воздействия вируса на различные звенья и взаимопотенцирующего влияния различных компонентов этих звеньев друг на друга.
На рис. 4 кратко представлены предлагаемые четыре основных патофизиологических механизма SARS-CoV-2, задействованных при повреждении ССС.
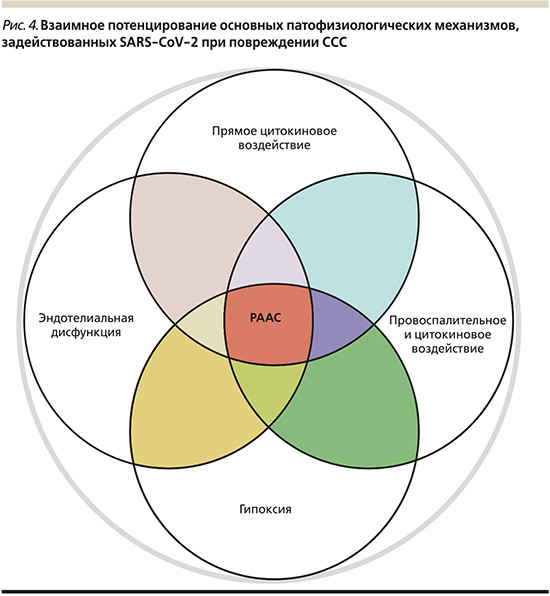
Центральным звеном развития сердечно-сосудистых осложнений, повреждения сердца и сосудов, формирования коагулопатии, по нашему мнению, является РААС. Помимо того, что специфическим рецептором для проникновения в клетку хозяина является АПФ-2, SARS-СoV-2 вызывает нарушение функционирования всех компонентов РААС. Дисбаланс прессорных и депрессорных механизмов, активация симпатической нервной системы и оксидативного стресса, задержка жидкости, запуск неконтролируемого апоптоза, увеличение провоспалительного и прокоагуляционного потенциала приводят в первую очередь к повреждению эндотелия кровеносных сосудов всех органов и систем. Необходимо напомнить, что в норме эндотелий сосудистого русла – активный эндокринный и паракринный орган, которому принадлежит ведущая роль в регуляции сосудистого тонуса (в первую очередь в микроциркуляторном русле) и гомеостаза. Нарушение функции эндотелия в свою очередь усугубляет дисбаланс прессорных и депрессорных систем, запускает каскад биологических реакций, активирующих апоптоз и потенцирующих коагуллопатию. В конечном итоге дисфункция эндотелия потенцирует дисбаланс РААС, запускает иммунопатогенетические воспалительные реакции с участием комплимента. Взаимопотенцирующее влияние РААС и поврежденного эндотелия проявляется резким ангиоспазмом, нарушением микроциркуляции, повреждением и гибелью клеток (прежде всего пневмоцитов II типа и эндотелиоцитов), что приводит к ОРДС, тромботическим осложнениям, ДВС-синдрому. Имеющиеся при COVID-19 проявления дыхательной гипоксии вносят свой весомый вклад в нарушение функционирование всех органов и систем и в совокупности с тканевой и гемической гипоксией оказывают мощное повреждающее воздействие на организм. Вновь активируются сигнальные пути РААС, еще больше усугубляется ЭД, что приводит к многократному усилению повреждающего действия. На этом фоне и без того напряженный иммунный ответ, обусловленный прямой вирусной токсичностью SARS-CoV-2, еще более «подстегивается» и характеризуется дисрегуляцией и гиперактивностью [19, 34]. Возникающий на этом фоне синдром высвобождения цитокинов способствует неконтролируемому дополнительному поражению организма, что в свою очередь оказывает потенцирующее действие на дисрегуляцию РААС, генерализацию ЭД.
Выводы
Несмотря на короткий опыт пандемии COVID-19, достигнуты значительные успехи в нашем нынешнем понимании сложного взаимопотенцирующего патогенеза повреждения ССС. Выявленные основные патогенетические механизмы являются теми терапевтическим целями, разумное воздействие на которые может ослабить, а в некоторых случаях и нивелировать негативное влияние вируса. Знание этих механизмов и их взаимо-связь позволяют нам использовать эти механизмы как основные направления для защиты ССС от повреждающего воздействия вируса. В первую очередь это относится к необходимости стабилизации РААС, препятствию проникновения вируса в клетку, минимизации проявлений ЭД, нормализации микроциркуляции и профилактики тромбообразования, борьбе с гипоксией (дыхательной, тканевой, гемической), снижению «напряжения» иммунитета и недопущению «цитокинового шторма». На настоящий момент отсутствуют клинические рекомендации по кардио- и вазопротекции при лечении CОVID-19. Также ввиду относительно недавнего начало пандемии нет законченных рандомизированных клинических исследований и мета-анализов, указывающих на неоспоримое превосходство тех или иных препаратов. Методы лечения для замедления репликации вируса и безу-словно разработка вакцин для профилактики имеют решающее значение. Тем не менее представляется, что для предотвращения или минимизации повреждения конечных органов необходим многогранный подход, направленный на решение некоторых или всех предлагаемых терапевтических задач.
Таким образом, лечение COVID-19 у пациентов кардиологического профиля, а также выбор стратегии по недопущению/минимизации повреждения сердца и сосудов носят индивидуальный характер с учетом возраста, пола, сопутствующих заболеваний и степени интенсивности инфекционного процесса, пока будущие исследования не откроют больше информации о патогенезе, а рандомизированные клинические испытания не укажут нам более структурированное направление для достижения наилучших результатов.


