
Термин «сахарный диабет» объединяет группу метаболических (обменных) заболеваний, характеризующихся хронической гипергликемией, которая является результатом нарушения секреции инсулина, действия инсулина или обоих этих факторов. Хроническая гипергликемия ведет к повреждению, нарушению функции и недостаточности жизненно важных систем и органов, что способствует ухудшению качества жизни и уменьшению продолжительности жизни.
Принципиальным отличием современной классификации сахарного диабета (СД), предложенной ВОЗ в 1999 г., служит ее этиологический принцип (см. таблицу) [1].
Авторы впервые предложили разделить этиологический фактор и степень дефицита инсулина, соответственно, четко выделить и разделить критерии, свидетельствующие об этиологии и дефиците инсулина. Так, под СД 1 типа (СД1) понимают такую форму заболевания, в основе которой лежит деструкция β-клеток, приводящая к дефициту инсулина. В большинстве случаев это происходит вследствие аутоиммунного процесса. Вместе с тем существует т.н. идиопатический СД1, также сопровождающийся развитием дефицита инсулина, этиология которого не до конца ясна. Данный вариант течения СД1 в большей степени характерен для лиц афро- и испаноамериканского происхождения. Данный тип СД упоминается в литературе под различными названиями: СД тип 1В, идиопатический СД, атипичный диабет, Flatbush диабет, а позднее СД 2 типа (СД2), склонный к кетозу [2]. Заболевание характеризуется острым началом, выраженной гипергликемией в дебюте заболевания, развитием кетоацидоза, однако затем наступает период ремиссии и оптимальный уровень гликемии поддерживается при соблюдении диеты или приеме пероральных сахароснижающих препаратов. При этом у пациентов отсутствуют маркеры аутоиммунного поражения β-клеток – антитела к антигенам β-клеток. Данный подтип СД в 2–3 раза чаще встречается среди мужчин среднего возраста с избыточной массой тела или ожирением.
Аутоантитела к антигенам β-клетки: к инсулину, ICA, GAD65, IA-2 и IA-2β, к транспортерам цинка являются общепринятыми маркерами аутоиммунной деструкции β-клеток. Данные антитела определяются среди 85–90 % пациентов с типичным СД1. Главной генетической детерминантой СД1 служат гены HLA-комплекса (DRB1, DQA1 и DQB (гаплотипы DR3 и DR4)), кодирующие белки, вовлеченные в иммунный ответ.
В типичных случаях дифференциальный диагноз СД1 и СД2 не представляет трудностей и основывается в основном на анализе клинической картины дебюта заболевания, возрасте больного, данных анамнеза. Так, для аутоиммунного СД1 характерны острое начало заболевания, часто отмечается связь с предшествующей вирусной инфекцией, молодой возраст пациента, выраженная гипергликемия, снижение массы тела в дебюте заболевания, развитие кетоацидоза. Единственным патогенетически правильным лечением остается инсулинотерапия. Быстро ухудшающееся состояние пациента в отсутствие инсулинотерапии может приводить к развитию комы.
Однако помимо типичных форм СД1 во взрослой популяции встречается особый вариант развития СД1: медленнопрогрессирующий аутоиммунный диабет взрослых – Latent autoimmune diabetes in adults (LADA) [3].
Первые работы, посвященные LADA, появились в средине 1980-х гг.; в них сообщалось о выявлении аутоанатител к антигенам β-клетки у больных с клинической картиной СД2, причем наличие аутоантител ассоциировалось с необходимостью раннего назначения инсулина для достижения компенсации углеводного обмена [4, 5]. По данным исследования UKPDS (United Kingdom Prospective Diabetes Study), аутоантитела к разным структурам β-клетки определялись у 12 % больных с впервые выявленным СД2. Частота выявления антител зависела от возраста дебюта заболевания: в группе от 25 до 34 лет антитела к GAD (глютаматдекарбоксилазе) были обнаружены у 34 % и к ICA (цитоплазматическому антигену β-клетки) у 21 % обследованных, в то время как в возрастной группе от 55–65 лет антитела к GAD регистрировались только у 7 % и к ICA у 4 % обследованных. При этом наличие антител служит значимым прогностическим фактором развития инсулинопотребности за первые годы заболевания. Так, 84 и 94 % обследованных в возрасте от 25 до 34 лет с положительными антителами к GAD и ICA были переведены на инсулин за первые 6 лет заболевания [6].
Частота выявления LADA колеблется от 3,8 до 10–15,0 % в различных этнических группах. Основные клинические особенности LADA: возраст дебюта заболевания старше 30 лет, клиническая картина СД2 в дебюте; компенсация углеводного обмена может быть достигнута соблюдением диеты или приемом пероральных сахароснижающих препаратов, однако уже за первые годы заболевания, в среднем через 6 лет, развивается потребность в инсулине.
Основные диагностические критерии LADA, которые позволяют рассматривать его как вариант течения СД1:
- присутствие аутоантител к GAD и/или ICA;
- низкий базальный и стимулированный уровень С-пептида;
- присутствие HLA аллелей высокого риска, характерных для СД1.
Важность проблемы своевременной диагностики LADA у взрослых заключается в следующем: исходная клиническая картина заболевания, «маскирующегося» под СД2, приводит к ошибочной диагностике и назначению пероральных сахароснижающих препаратов, вместе с тем основной патогенетически правильной тактикой лечения LADA остается инсулинотерапия. Представляют интерес результаты Кокрановского мета-анализа публикаций, представленных в электронных базах MEDLINE, EMBASE и посвященных проблеме выбора тактики лечения LADA [7]. Для анализа было отобрано 7 исследований, которые включали в общей сложности 735 участников. Публикации низкого качества представления материала и исследования с небольшим количеством участников не рассматривались. Длительность отобранных исследований составляла от 6 месяцев до 10 лет. Ни в одном из исследований не были представлены данные о применении метформина или глитазонов в виде монотерапии данных больных. Ни росиглитазон, ни производные сульфонилмочевины (ПСМ) в комбинации с инсулином не имели преимуществ по сравнению с терапией только инсулином в отношении улучшения метаболического контроля. Только в одном исследовании было показано, что стимулированный уровень С-пептида остается более высоким при терапии инсулином по сравнению с ПСМ (среднее различие – 7,7 нг/мл (95 % CI – 2,9–12,5). Проведенный анализ позволил сделать следующие выводы. На сегодняшний день исследований, посвященных лечению и возможной профилактике LADA, мало. Существующие же имели высокий риск предвзятого суждения и были проведены в разных этнических группах (Китай, Япония, Куба, Великобритания, Швеция). Очевидно, что ПСМ не должны использоваться в качестве препаратов первой линии в лечении больных СД2 с положительными антителами; патогенетически правильным является проведение инсулинотерапии. В отношении других видов терапии нет доказательств существенных преимуществ.
Знание особенностей дебюта СД1 в различных возрастных и этнических группах позволяет сделать заключение о необходимости использования при диагностике иммуногенетических методов исследований с целью уточнения диагноза и начала патогенетического лечения.

Помимо СД1 и СД2 все другие типы СД в современной классификации объединены в группу «Другие специфические типы СД». Именно эти типы СД, а также нетипичные случаи СД1, такие как LADA и идиопатический СД, не диагностируются вовремя, что приводит в свою очередь к ошибкам при выборе тактики лечения. Вместе с тем постановка правильного диагноза не только чрезвычайно важна для определения лечения, но и позволяет предсказывать клиническое течение заболевания, объяснять другие связанные с заболеванием клинические проявления, в ряде случаев – определять тактику лечения заболевших родственников.
Среди специфических типов СД большую группу составляют больные, у которых СД развился на фоне или вследствие поражения экзокринной части поджелудочной железы. Это могут быть панкреатит (острый и хронический), травма, панкреатэктомия, опухоли поджелудочной железы, муковисцидоз, гемохроматоз, фиброкалькулезная панкреатопатия и др. [8, 9]. По данным статистики США и Европы, СД на фоне панкреатита составляет около 1 % всех его случаев.
Особенности СД после панкреатэктомии и операций на поджелудочной железе:
- сосуществование абсолютного дефицита инсулина и дефицита глюкагона;
- наличие мальабсорбции в связи с внешнесекреторной недостаточностью;
- низкая потребность в инсулине при высокой чувствительности к нему;
- склонность к гипогликемиям (глюконеогенез на низком уровне в связи с недостатком глюкагона);
- редкое развитие кетоацидоза.
У больных с хроническим панкреатитом степень нарушения углеводного обмена колеблется в широких пределах: от нарушения толерантности к глюкозе до инсулинопотребного СД. При хроническом панкреатите на ранних стадиях заболевания уровень инсулина нормальный или умеренно повышен, уровень глюкагона в пределах нормы, с прогрессированием заболевания наблюдается атрофия островковых клеток, замещение их соединительной тканью, что приводит к развитию гипоинсулинемии и гипоглюкагонемия.
Клиническое течение СД у больных хроническим панкреатитом имеет свои особенности: пациенты чаще нормального или худощавого телосложения; нет связи с семейной предрасположенностью, ожирением, инсулинорезистентностью. Cимптомы диабета обычно появляются спустя несколько лет после начала болевых приступов [10]. Сахарный диабет при панкреатите, в особенности в начале заболевания, протекает легче «эссенциального». Патогенетическим лечением считается инсулинотерапия, однако потребность в инсулине обычно сравнительно невысока, редко развивается кетонурия и диабетическая кома. В результате длительно продолжающегося хронического бессимптомного панкреатита вторичный СД развивается приблизительно в 5 % случаев в отсутствие дальнейших обострений [11]. Однако в случае хронического рецидивирующего панкреатита через 20 лет 25–30 % пациентов имеют нарушение толерантности к глюкозе, у 40–50 % развивается СД. Было замечено, что при некальцифицирующем хроническом панкреатите частота случаев нарушения толерантности к глюкозе составляет 50 %, а у 30 % больных развивается СД. При кальцифицирующем хроническом панкреатите эти показатели выше и составляют соответственно 90 и 61 %. Отличие острого, острого рецидивирующего и хронического рецидивирующего панкреатита (острой атаки хронического панкреатита) состоит в том, что после острого панкреатита возможно полное восстановление ткани поджелудочной железы. Хронический рецидивирующий панкреатит сопровождается остаточными структурно-функциональными нарушениями. Однако даже при наличии СД на фоне хронического панкреатита уровень С-пептида никогда не достигает низких значений, наблюдаемых при СД1.
Сахарный диабет 1 и 2 типов относится к полигенным мультифакторным заболеваниям, т.е. в их основе лежит наличие определенных комбинаций неблагоприятных аллельных вариантов нескольких генов, что на фоне провоцирующих факторов внешней среды приводит к манифестации заболевания. На долю редких моногенных вариантов СД приходится не более 1 % всех случаев СД. К моногенным формам СД относятся различные формы MODY (Maturity Onset Diabetes for Young) диабета, неонатальный СД (транзиторный и перманентный), СД, диабет, развивающийся при мутациях митохондриальной ДНК. Для подтверждения диагноза необходимо генетическое тестирование.
В отсутствие типичных для СД1 и СД2 симптомов моногенные формы СД можно заподозрить при наличии следующих признаков [12]:
- СД, развившиеся в течение первых 6 месяцев жизни;
- аутосомно-доминантный тип наследования СД;
- умеренная гипергликемия натощак (5,5–8,5 ммоль/л), особенно в раннем возрасте и в случае семейного заболевания;
- сочетание СД с экстрапанкреатическими проявлениями: нейросенсорной тугоухостью, атрофий зрительного нерва, пигментной дегенерацией сетчатки, заболеванием печени, почек, другими синдромальными проявлениями;
- признаки секреции эндогенного инсулина: определяемый С-пептид более чем через 3 года от дебюта заболевания при уровне глюкозы более 8 ммоль/л.
Неонатальный СД развивается в первые 6 месяцев жизни и встречается с частотой от 1/300 тыс. до 1/500 тыс. новорожденных [13]. Антитела к антигенам β-клетки, ассоциация с аллелями высокого риска развития СД1 отсутствуют. Выделяют перманентную и транзиторную формы неонатального СД. При транзиторном неонатальном СД (ТНСД) инсулинотерапия требуется в течение первых нескольких месяцев жизни, но менее чем через 18 месяцев наступает ремиссия заболевания. В 50 % случаев наблюдается рецидив, чаще в подростковом возрасте или во взрослом состоянии. Перманентный неонатальный СД (ПНСД) протекает на протяжении всей жизни. Наиболее частой причиной ТНСД остатся нарушение импринтинга генов ZAC/HIAMI на локусе 6q24. Неонатальный СД может вызываться экспрессией мутантных генов, нарушающих основные клеточные процессы. В некоторых случаях как ТНСД, так и ПНСД могут быть связаны с мутацией генов, контролирующих АТФ-зависимые калиевые каналы – KCNJ11 и ABCC8. В связи с этим в ряде исследований последних лет была установлена эффективность терапии неонатального СД препаратами сульфонилмочевины в дозах, превышающих рекомендуемые для лечения СД2. Кроме основных двух форм неонатального СД было идентифицировано множество клинических синдромов, связанных с ПНСД: IPEX-синдром, синдром Уолкотта–Роллисона, сочетание ПНСД с гипоплазий и атрофией поджелудочной железы. Идентификация генетических мутаций становится важным инструментом в арсенале клинициста, помогая обеспечить точную диагностику, адекватное лечение и высокий уровень медико-генетического консультирования.
Наследуемые по материнской линии диабет и глухота (MIDD – maternally inherited diabetes and deafness) – мультиорганные заболевания, проявляющиеся СД, нейросенсорной глухотой и макулярной дистрофией сетчатки [14, 15]. Относится к митохондриальным заболеваниям и может поражать как мужчин, так и женщин, но передается исключительно по материнской линии, поскольку митохондриальная ДНК происходит из яйцеклетки. Митохондриальные болезни обусловлены генетическими и структурно-биохимическими дефектами митохондрий и сопровождаются нарушением тканевого дыхания. В связи с возникающим системным дефектом энергетического метаболизма поражаются в различных комбинациях наиболее энергозависимые органы и ткани-мишени (мозг, скелетные мышцы, миокард, орган зрения, поджелудочная железа, печень, почки).
Чаще СД в составе синдрома MIDD имеет относительно ранний возраст начала (до 35 лет); масса тела бывает нормальной или сниженной. Также характерным для синдрома MIDD является наличие двусторонней нейросенсорной глухоты и макулярной дистрофии сетчатки с линейной пигментацией вокруг макулы и диска зрительного нерва. Митохондриальный диабет может проявляться как СД1, так и СД2. В случае возникновения диабета у лиц, фенотипически схожих с пациентами СД2, возможно ведение пациента на диетотерапии и препаратах сульфонилмочевины. Метформин не показан из-за высокой склонности и риска развития лактат-ацидоза. Однако недостаток инсулина в большей части случаев со временем прогрессирует и через несколько лет от дебюта заболевания возникает потребность в инсулине.
Для подтверждения диагноза необходимо исследование митохондриальной ДНК, выделенной из клеток крови на носительство конкретных мутаций у больных с четким фенотипом. Альтернативной возможностью молекулярной диагностики может стать идентификация конкретного биохимического дефекта в том или ином звене дыхательной цепи митохондрий.
С расширением диагностических возможностей генетических методов исследования стала доступной диагностика семейных форм СД, ранее относимых к СД1 или СД2. Среди форм наследственного СД или MODY наиболее часты мутации гена HNF-1-альфа (MODY-3), HNF-4-альфа (MODY-1), гена глюкокиназы (MODY-2).
MODY-3 – наиболее частый вариант в данной подгруппе и характеризуется началом в раннем возрасте (4–18 лет), выраженной постпрандиальной гипергликемией, сниженным почечным порогом, в связи с чем глюкозурия может определяться при нормоглокемии [16]. Заболевание прогрессирует с возрастом. Отмечается выраженная чувствительность в ПСМ.
MODY-1 (мутации гена HNF-4α) во многом похож на MODY-3. СД вследствие мутации гена глюкокиназы (MODY-2) сложен для диагностики из-за скудности проявлений. Наблюдается легкая гипергликемия натощак (5,5–8,5 ммоль/л), при нормальном уровне гликогемоглобина (НbА1с) (5,5–5,7 %). Состояние не прогрессирует, редко развиваются осложнения. При обследовании ближайших родственников часто удается выявить и у них схожие нарушения углеводного обмена.
MODY-5 первоначально рассматривался как изолированный СД, однако позже была установлена ассоциация с такими врожденными аномалиями, как аномалии развития почек и мочевыводящих путей, гениталий, поджелудочной железы, печени [17]. Причина в мутации гена TCF2, кодирующего ядерный фактор гепатоцитов-1β.
В настоящее время MODY-5 вынесен из классификации СД и рассматривается как отдельный генетический синдром. Сахарный диабет при этом характеризуется тяжелым течением, показана инсулинотерапия. Сахарный диабет может присутствовать в составе других генетических синдромов, таких как синдром Дауна, Клайнфельтера, Тернера, Прадера–Вилли, Лоуренса-Муна–Барде–Бидля, атаксии Фридрейха и др.
Описаны редкие генетические синдромы, при которых мутации генов рецепторов инсулина стали причиной развития выраженной инсулинорезистенности, например лепречаунизм, синдром Рабсона–Менденхолла, инсулинорезистентность типа А, липоатрофический СД. Общим для всех проявлений служат повышенный уровень инсулина в плазме крови в отсутствие ожирения, повышение уровня андрогенов, наличие acantosis nigricans.
Нарушения углеводного обмена при эндокринопатиях могут возникать при следующих ситуациях: существование двух независимых заболеваний – СД и другого эндокринного заболевания и нарушение углеводного обмена вследствие эндокринного заболевания. Довольно часто встречается умеренная степень нарушений углеводного обмена – нарушение толерантности к глюкозе или СД, который удается компенсировать диетой. Однако бывают и более выраженные нарушения. В большинстве случаев наблюдается усиление инсулинорезистентности вследствие продукции контринсулярных гормонов, как, например, при синдроме Кушинга.
В 90 % всех случаев СД взрослых составляет СД2. Исследования последних десятилетий во многом расширили наше понимание патогенеза СД2. Несомненным остается наличие резистентности мышечной, жировой ткани, а также ткани печени к действию инсулина. Инсулинорезистентность тканей предшествует развитию СД и находится под влиянием генетических и факторов внешней среды (образ жизни, характер питания). Главным нарушением, приводящим к манифестации СД2, является нарушение секреции инсулина. Открыт инкретиновый механизм регуляции секреции инсулина и доказано его нарушение при СД2. Показано увеличение уровня глюкагона у больных СД2, в связи с чем разрабатываются препараты – антагонисты рецепторов глюкагона. Изучение механизмов реабсорбции глюкозы в почках привело к созданию нового класса сахароснижающих препаратов – ингибиторов натрий-глюкозного транспортера SGLT-2. Было показано, что инсулин способен проникать в гипоталамус и оказывать влияние на секрецию нейропептида Y и проопиомеланокортина. Действие инсулина опосредовано через центральные АТФ-зависимые К+-каналы и активацию автономной нервной системы. В 2010 г. препарат центрального действия бромкрептин был одобрен FDA для лечения СД2 в виде монотерапии. Его действие направлено на восполнение дофамина, недостаток которого приводит к активации симпатической нервной системы, гиперпродукции глюкозы печенью, липолизу, резистентности к инсулину.
Сегодня в арсенале диабетолога имеется широкий спектр лекарственных препаратов для коррекции гипергликемии, множество препаратов проходит стадии клинических исследований. Все это указывает на необходимость четких рекомендаций по лечению СД2.
Общим международным руководством для создания локальных алгоритмов лечения СД2 является алгоритм Международной федерации диабета (IDF) 2011 г. Изменения образа жизни, включающие модификацию диеты, повышение уровня физической активности, коррекцию избыточной массы тела и отказ от курения, составляют основу лечения СД2 [18]. Для большинства больных определена цель лечения – достижение уровня гликированного гемоглобина (НbА1с) < 7,0 %, однако подчеркивается, что целевые значения должны быть индивидуализированы. Последующие изменения в лечении производят, если через 3 месяца целевой уровень НbА1с не достигнут. На каждом последующем этапе в руководстве IDF предлагается как обычный, так и альтернативный подход. В качестве препарата первого выбора рекомендован метформин при условии отсутствия к нему противопоказаний, таких, например, как почечная недостаточность, и удовлетворительной переносимости препарата. Основанием этого предложения стали такие свойства препарата, как благоприятное его влияние на массу тела, отсутствие гипогликемических состояний, благоприятное влияние на сердечно-сосудистую систему и невысокая стоимость. В качестве альтернативных препаратов первого ряда в алгоритме IDF рассматриваются ПСМ и ингибиторы α-глюкозидазы. Если монотерапия не позволяет достигать целевого уровня гликемии, необходимо назначать второй препарат: в качестве стандартного подхода пациентам на метформине рекомендуется добавлять ПСМ или (альтернативный вариант) ингибитор α-глюкозидазы, ингибитор дипептидилпептидазы-4 (ДПП-4) или тиазолидиндион. Если контроль диабета все еще остается неудовлетворительным, стандартный подход, согласно алгоритму IDF, заключается в добавлении третьего перорального препарата либо переводе пациента на инсулинотерапию. Агонисты рецепторов глюкагоноподобного пептида-1 (ГПП-1) указываются только как препараты альтернативного подхода – в основном в связи с их стоимостью.
Положения Американской диабетологической ассоциации (ADA) и Европейской ассоциации по изучению СД (EASD) в отношении тактики лечения СД2 [19] претерпели в 2012 г. значительные изменения. Были внесены следующие основные уточнения: рекомендовано начинать терапию пероральными сахароснижающими препаратами сразу в момент постановки диагноза в комбинации с модификацией образа жизни и снижением веса; определены цели лечения – достижение гликированного гемоглобина для большинства больных менее 7 %; оценка эффективности и изменение терапии должны проводиться каждые 3 месяца; начальным препаратом остается метформин при условии отсутствия противопоказаний и хорошей переносимости препарата.
При неэффективности монотерапии в качестве второго препарата для комбинированной терапии могут быть препараты следующих групп: ингибиторов ДПП-4, ПСМ, тиазолидиндионы, аналоги ГПП-1, инсулин (базальный). В рекомендациях отмечается, что в настоящее время недостаточно данных в отношении преимуществ тех или иных препаратов при следующем этапе лечения. Разумна комбинация метформина с 1–2 сахароснижающими пероральными или инъекционными препаратами с учетом возможных побочных эффектов и противопоказаний. Основные факторы, определяющие выбор препарата: его сахароснижающая способность, риск развития гипогликемии, влияние на массу тела, побочные эффекты и стоимость. Однако всегда ли этого достаточно для эффективного лечения?
На 49-м ежегодном Конгрессе Европейской ассоциации по изучению СД (EASD) 2013 г. ведущий мировой лидер диабетологии Stefano Del Prato выступил с докладом «Treatment of type 2 diabetes based on pathophysiological knowledge», в котором наглядно доказал, что лечение СД2 должно базироваться на понимании патогенеза СД2, в основе которого помимо инсулинорезистентности лежит прогрессирующее ухудшение структуры и функции β-клеток. В связи с этим терапия СД должна быть направлена как на улучшение усвоения глюкозы тканями, уменьшение глюконеогенеза, так в значительной степени на сохранение массы и функции β-клеток, а возможно, и на их восстановление.
Но СД2 также неоднороден. И инсулинорезистентность, и нарушение секреции инсулина в каждом конкретном клиническом случае могут быть выражены в разной степени и прогрессировать разными темпами. Наглядным примером индивидуального подхода к лечению больных с первоначальным диагнозом СД2 служат результаты датского исследования, также представленного на Конгрессе EASD (2013). Henning Beck-Nielsen представил доклад «Phenotypes in type 2 diabetes based on a national survey». Целью последнего было выделить патофизиологические фенотипы СД2. Всего были обследованы 1048 пациентов с впервые выявленным СД2. Средний возраст составил 61 год [53, 67]. Все пациенты были направлены к эндокринологу врачами общей практики с первоначальным диагнозом «СД 2 типа». После осмотра эндокринологом и проведения иммунологических исследований было установлено, что у 3 % обследованных больных определены антитела к GAD, причем диагностически значимым считался титр GAD ≥ 20. Следовательно, такие пациенты рассматривались как имеющие в дальнейшем медленнопрогрессирующий аутоиммунный СД (LADA). После подробного сбора анамнеза и при необходимости определения уровня липазы в крови у 3,9 % больных был диагностирован СД вследствие панкреатита. У 5,8 % СД был связан с терапией стероидами. Редкие формы СД составили 0,6 %.
Таким образом, истинный СД2 наблюдался только у 86,7 % больных. Однако исследователи поставили следующую задачу: может ли классический СД2 быть разделен на патофизиологические субфенотипы? Всем больным истинным СД2 была проведена оценка функции β-клеток и чувствительности к инсулину с помощью HOMA-модели (HOMA2Beta и HOMA2S).
На основании полученных данных СД2 был разделен на три патофизиологических фенотипа:
- классический СД2: умеренная инсулинорезистентность и относительное снижение функции β-клеток;
- инсулинопенический: нормальная или повышенная чувствительность к инсулину и абсолютная недостаточность β-клеток;
- гиперинсулинемический: выраженная инсулинорезистентность и увеличение функции β-клеток.
Пациенты выделенных трех групп различались и по своему фенотипу. Наибольший индекс массы тела и обхват талии имели пациенты с гиперинсулинемическим типом секреторного ответа. Интересно, что именно среди пациентов этой группы 9,6 % уже имели перенесенный инфаркт миокарда в анамнезе. Самый низкий индекс массы тела и охват талии, а также число перенесенных инфарктов миокарда в анамнезе наблюдались у больных с инсулинопеническим типом секреции инсулина.
Таким образом, результаты исследования наглядно продемонстрировали фенотипическую и патогенетическую неоднородность СД2, что, несомненно, указывает и на необходимость дифференциального, индивидуального подхода к лечению.
Нарушение секреции инсулина – основное звено в патогенезе СД2.
При этом наблюдаются следующие нарушения функции β-клеток:
- снижение или потеря первой фазы секреции инсулина;
- удлинение 2-й фазы стимулированной секреции инсулина;
- снижение или неадекватность секреции инсулина;
- изменения в осцилляторной секреции инсулина;
- повышение секреции проинсулина.
Производные сульфонилмочевины являются наиболее часто используемыми сахароснижающими препаратами во всем мире. Их сахароснижающий эффект обусловлен стимуляцией секреции инсулина. Эти препараты эффективны, только когда имеется достаточное количество функционально активных β-клеток. Сахароснижающий эффект ПСМ связан с активацией АТФ-зависимых калиевых каналов (КATP-каналы). Их стимулирующее влияние на секрецию и высвобождение инсулина усиливается в присутствии глюкозы. Обеспечивая изменение мембранного потенциал, КATP-каналы участвуют в регуляции обменных процессов в разных тканях, включая β-клетки, сердце, скелетные мышцы, гладкомышечную ткань сосудов, ткань мозга [20, 21]. КATP-каналы имеют тканевую специфичность, что зависит от структуры и молекулярной массы составляющих субъединиц, что в свою очередь определяет электрофизиологические свойства тканей, их реакцию на различные факторы и лекарственные препараты. Производные сульфонилмочевины по-разному взаимодействуют с АТФ-зависимыми К-каналами β-клетки (SUR – рецепторы сульфонилмочевины). Было показано, что гликлазид модифицированного высвобождения (МВ) обратимо взаимодействует с АТФ-зависимыми калиевыми каналами β-клетки и не связывается с данным рецептором в других тканях, в частности на кардиомиоците. Блокада КATP-каналов сердца препаратами может стать пагубной для миокарда в состоянии ишемии за счет подавления ишемической предподготовки. Ряд препаратов, ПСМ (глибенкламид и толбутамид) связываются с КАТФ-каналами как β-клетки, так и кардиомиоцита. По той же причине глибенкламид упраздняет кардиопротективный эффект ишемической предподготовки за счет закрытия калиевых каналов, что может приводить к усилению повреждения миокарда в условиях ишемии и увеличению площади инфаркта [22]. Таким образом, можно говорить о высокой селективности действия гликлазида МВ, что свидетельствует о преимуществе назначения данного препарата больным СД2 с ИБС, особенно при ее осложненном течении.
Обратимость взаимодействия препарата с рецептором объясняет низкий риск гипогликемических состояний при его применении. Известно, что 90 % больных группы интенсивного контроля в исследовании ADVANCE (Action in Diabetes and Vascular disease; Preterax and Diamicron MR Controlled Evaluation) принимали Диабетон МВ, причем 70 % из них в дозировке 120 мг/сут [23]. При этом в данной группе пациентов количество эпизодов гипогликемии было в 7 раз меньше, чем в группе интенсивного контроля исследования ACCORD (Action to Control Cardiovascular Risk in Diabetes), и в 2 раза меньше, чем в исследовании UKPDS. Более того, в течение 5 лет стратегия интенсивного контроля гликемии, основанная на использовании Диабетона MВ, не приводила пациентов с СД2 к увеличению массы тела. Риск гипогликемий при лечении Диабетоном МВ был низким и составил менее 5 % [24].
Допустимо применение Диабетона МВ при диабетической нефропатии на стадии протеинурии при скорости клубочковой фильтрации не ниже 30 мл/мин.
Анализ комбинированного влияния интенсивного контроля гликемии, основанного на применении гликлазида МВ, и гипотензивной терапии на макро- и микрососудистые осложнения в самом масштабном в диабетологии исследовании ADVANCE показал неоспоримую эффективность и необходимость многофакторного подхода к профилактике осложнений СД и подтвердил статистически значимое снижение риска как микро-, так и макроальбуминурии. Результаты исследования были столь убедительны, что позволили сделать дополнение к инструкции по применению препарата гликлазид МВ, согласно которому этот препарат рекомендуется теперь в качестве средства профилактики осложнений СД, для снижения риска микро- (нефропатия, ретинопатия) и макрососудистых (инфаркт миокарда, инсульт) осложнений у пациентов с СД2 путем интенсивного контроля гликемии.
Сегодня активно обсуждается вопрос о том, что длительное применение ПСМ может приводить к дисфункции и апоптозу β-клеток, тем самым ускоряя развитие потребности в инсулине. Прогрессирующая недостаточность β-клеток лежит в основе естественного течения СД2. Уже в дебюте заболевания функция β-клеток снижена на 50 %.
Примерно в той же степени снижена и масса β-клеток. Эти процессы во многом обусловлены генетическими факторами, нарушениями внутриутробного развития поджелудочной железы, а также воздействием таких вторичных факторов, как глюкозотоксичность и липотоксичность. Хроническая гипергликемия (глюкозотоксичность) приводит к усилению выработки цитокина IL-1β, развитию оксидативного стресса, влияет на экспрессию Fas-рецептора и Fas-лигандов, активирует процессы апоптоза [25]. Предполагается, что стойкое воздействие высоких уровней глюкозы и уровней реактивных форм кислорода на β-клетки вызывает дефицит факторов транскрипции, необходимых для поддержания нормальных уровней активности промотора гена инсулина и приводящих к уменьшению секреции инсулина. Наряду с этим в нескольких исследованиях отмечено, что увеличение поступления ионов Ca2+ в клетку вследствие применения ПСМ остается одной из причин гибели β-клеток [26, 27].
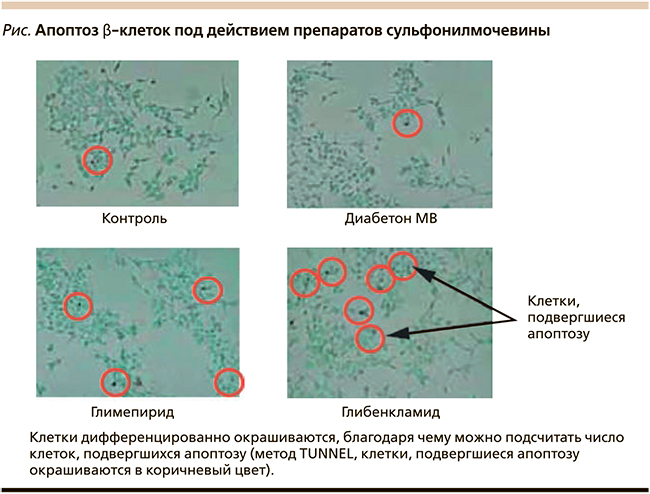
Однако до конца не известно: имеются ли различия между ПСМ в отношении влияния на β-клетку? В связи с этим представляют интерес результаты исследования, в котором оценивалось влияние различных ПСМ и натеглинида на уровни окислительного стресса и апоптоза в культуре панкреатических β-клеток линии MIN6 [28]. После культивирования клетки MIN6 подвергали воздействию препаратов сульфонилмочевины (глибенкламида, глимепирида и гликлазида) и натеглинида в различных концентрациях. Воздействие на клетки MIN6 глибенкламида, глимепирида и натеглинида в течение 24 часов привело к значительному увеличению внутриклеточного образования реактивных форм кислорода, величина которого зависела от концентрации препарата. Судя по результатам оценки образования реактивных форм кислорода, стимулирующие эффекты глибенкламида на апоптоз были достоверно выше, чем эффекты глимемирида или натеглинида. В отличие от этого обработка клеток гликлазидом не привела к достоверному увеличению числа апоптозных клеток (см. рисунок).
Таким образом, ПСМ имеют многолетний опыт применения. Препараты данного класса неоднородны по своим свойствам. При этом гликлазид МВ отвечает всем требованиям, предъявляемым сегодня к современным сахароснижающим препаратам для лечения СД2: не влияет на процессы апоптоза β-клеток, имеет низкий риск развития гипогликемических состояний, практически не влияет на массу тела, является безопасным для лиц с сердечно-сосудистыми заболеваниями и может применяться больными диабетической нефропатией.


