
Атопический дерматит (атопическая экзема, синдром атопической экземы/дерматита; в МКБ-10: раздел L20) – хроническое воспалительное заболевание кожи, сопровождающееся зудом, возрастной морфологией высыпаний, стадийностью, частым инфицированием. Как правило, атопический дерматит (АтД) начинается в раннем детском возрасте, может продолжаться или рецидивировать в зрелом возрасте и приводит к существенному ухудшению качества жизни как самого больного, так и членов его семьи. Поэтому проблема своевременной и адекватной терапии заболевания в настоящее время приобрела особую медицинскую и социальную значимость. АтД в большинстве случаев развивается у лиц с наследственной предрасположенностью и часто сочетается с другими аллергическими болезнями, такими как бронхиальная астма (БА), аллергический ринит (АР), пищевая аллергия, а также с рецидивирующими кожными инфекциями. Основными органами-мишенями при атопических заболеваниях являются барьерные ткани – кожа, легкие, верхние дыхательные пути и кишечник. В то время как для аллергических болезней дыхательных путей характерным механизмом являются аллергические реакции I типа, при АтД наблюдается значительно более сложный по патогенезу хронический воспалительный процесс, локализующийся в коже.
Основные представления о патогенезе
АтД рассматривается в настоящее время как болезнь со сложным патогенезом, с генетической предрасположенностью, которая обусловливает особенности врожденного и адаптивного иммунного ответа, включая взаимодействие макроорганизма с факторами окружающей среды (воздействие аллергенов, ирритантов, микроорганизмов, продуктов питания). Нарушения кожного барьера при АтД, повышение его проницаемости могут способствовать трансдермальному проникновению аллергенов окружающей среды и системной сенсибилизации аллергенами, что объясняет частую его ассоциацию с пищевой аллергией, БА, АР.
Несмотря на то что АтД обычно представляется как единое заболевание, недавние исследования показывают, что целесообразно выделение различных фено- и эндотипов болезни, аналогично тому, как в настоящее время пытаются классифицировать БА и риносинусит, основываясь на совокупности дебюта болезни, биомаркеров, иммунной поляризации, генных вариантов и естественной истории заболеваний [1].
Идентификация иммунных особенностей при АтД имеет особое значение, т.к. могут быть определены показания к применению биологических методов терапии, направленных на коррекцию конкретных иммунных патологических механизмов, ассоциируемых, например, с Th2-вариантом иммунного ответа, с преобладанием тех или иных воспалительных цитокинов и медиаторов, задействованных в патогенезе болезни.
Особое внимание в последние годы уделяется также влиянию на течение АтД дисфункций эпителиального кожного барьера, как генетически детерминированных, так и приобретенных. Существует мнение, будто раннее начало применения эмолентов (средства/крем, смягчающие кожу) при ведении пациентов с АтД способно редуцировать его симптомы и даже может быть рассмотрено как профилактика развития болезни и ее прогрессирования [2].
В настоящее время рассматриваются две концепции формирования дисфункции кожного барьера при АтД:
- «Внешняя» («outside-in hypothesis»), согласно которой основой нарушения эпидермального барьера являются его генетические дефекты, приводящие к повышению кожной проницаемости, трансдермальному проникновению аллергенов и колонизации кожи микроорганизмами.
- «Внутренняя» («inside-out hypothesis») – в данном случае в основе формирования дефектов кожного барьера лежат изменения иммунного ответа, что приводит к вовлечению дермы в процесс иммунного воспаления и неизбежно отрицательному влиянию на проницаемость эпидермального барьера.
- «Смешанная» – существует также мнение, будто оба процесса могут быть задействованы в отношении больных АтД, т.к. у большинства пациентов имеет место комплекс генетических дефектов эпидермального барьера и измененных иммунных реакций в ответ на воздействие факторов окружающей среды.
Эффективная профилактика и лечение АтД требуют многостороннего подхода, учитывающего восстановление и поддержание целостности эпидермального барьера и его гидролипидной мантии, контроль воспаления кожи, подбор питания, идентификацию аллергенных и микробных триггеров, купирование их воздействия, снижение трансдермальной потери влаги. Установлено, что для части пациентов с АтД характерны мутации гена, кодирующего синтез филаггрина – структурного белка кожи, который специфически взаимодействует с промежуточными филаментами – кератинами. Образуется филаггрин из профилаггрина – нерастворимого полипротеина, который в результате дефосфорилирования и разложения образует мономеры филаггрина в роговом слое кожи. Профилаггрин представляет собой основной компонент кератогиалиновых гранул, видных в световой микроскоп в зернистом слое эпидермиса. Мономерный филаггрин связывается с кератином и другими промежуточными белками – филаментами кератинового цитоскелета, образуя тесные связи между этими волокнами. Таким образом, происходит коллапс и уплощение клеток на поверхности рогового слоя с образованием чешуек (у млекопитающих на кератиновые волокна и связывающий их филаггрин приходится 80–90% общей массы белка эпидермиса). В конечном счете филаггрин в верхней части рогового слоя распадается на отдельные аминокислоты. Филаггрин содержит массу гистидина, который метаболизируется до органических кислот, помогающих поддерживать в эпидермисе требуемое значение градиента рН. Мутации в гене филаггрина (FLG) с последующей потерей его функции проявляются в нарушении барьерных функций эпидермиса, развитии АтД и аллергической сенсибилизации.
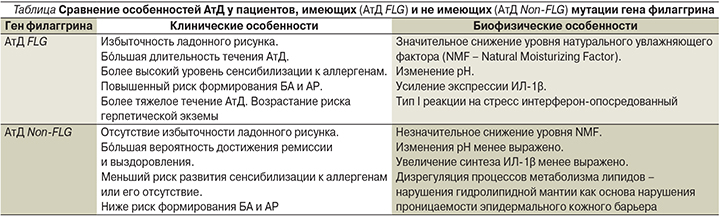
К характерным симптомам, сопровождающим нарушения метаболизма филаггрина, относятся сухость кожи, раннее начало АтД, течение которого торпидно и часто сочетается с БА, пищевой аллергией и инфицированием кожи [3]. Последние исследования показывают, что пациенты, имеющие мутации гена, отвечающего за синтез филаггрина, имеют клинические и патогенетические особенности, отличающие их от пациентов с АтД, не имеющих данных мутаций (см. таблицу). Пациенты с мутациями, приводящими к потере функции филаггрина, демонстрируют усиление экспрессии интерлейкина-1 (ИЛ-1) в роговом слое кожи, и первый тип реакции на стресс – интерферон-опосредованный [4]. Для детей с АтД, имеющих нормальные гены филаггрина (FLG), больше свойственны процессы нарушения регуляции липидного обмена, нарушения со стороны гидролипидной мантии кожи, повышение трансдермальной проницаемости при данном варианте болезни.
Исследования показали, что носительство делеции 2282del4 в гене FLG значимо повышает риск развития АтД в российской популяции [5]. Установлено, что наличие мутации 2282del14 в гене FLG как в гетерозиготном, так и в гомозиготном состоянии является риском развития непрерывно рецидивирующего среднетяжелого и тяжелого течения АтД в сочетании с другими аллергическими заболеваниями (БА, круглогодичным персистирующим АР, поллинозом) [6].
Установлено также, что филаггрин влияет на секрецию сфингомиелиназы, которая участвует в защите от стафилококкового α-токсина, вызывающего гибель кератиноцитов, и способствует колонизации кожи стафилококками [7].
В связи с этим предлагается рассматривать пациентов с мутациями гена FLG как особый подтип больных АтД с последующим формированием специфических для данного подтипа больных подходов к терапии, в основе которых лежит восстановление барьерной функции кожи. Частота встречаемости мутаций гена FLG зависит от популяции. Тем не менее данные мутации встречаются среди 40% больных детей с тяжелым течением АтД [8].
Таким образом, недавно выявленная связь между мутациями в гене FLG с риском развития и степенью тяжести АтД показывает важность комплекса эпидермальной дифференцировки при данной патологии и должна быть учтена при построении терапевтических стратегий.
Кроме гена FLG АтД связан с вариациями в других генах, которые кодируют кластер белков в комплексе эпидермальной дифференцировки, расположенном в хромосоме 1q21. Они включают, в частности, хорнерин и предшественник рогового конверта SPRR3. Однако в отличие от филаггрина их биологическое значение по отношению к АтД пока еще не очень хорошо понятно. Известно, однако, что дисфункции эпидермального барьера могут вызывать аллергические заболевания кожи. Вариативность в генах эпидермиса и генах, контролирующих врожденные и адаптивные иммунные ответы, в комплексе с особенностями воздействия окружающей среды в конечном итоге, по-видимому, и формируют клиническую и патогенетическую картину болезни.
Если роговой слой нарушен, например, из-за дефицита таких структурных белков, как филаггрин, инволюкрин, лорикрин, и/или липидов, таких как церамиды, компенсаторно для сохранения функции могут быть привлечены другие барьерные структуры. Они включают жесткие белки перехода, такие как клаудинс, которые находятся на противоположных мембранах зернистого слоя кератиноцитов непосредственно под роговым слоем и тем самым образуют второй физический барьер в эпидермисе.
Нормальная кожа может рассматриваться как последовательность взаимосвязанных барьеров, функции которых заключаются в удержании влаги и отражении трансдермального проникновения антигенов, включая аллергены и инфекционные инвазии. Если физические барьеры нарушены, наблюдается инициирование реакций со стороны структур врожденного иммунитета, чтобы предотвратить дальнейшее вторжение антигенов. Кератиноциты и антиген-представляющие клетки, расположенные в коже, через врожденные рецепторы распознавания образов, такие как Toll-подобные рецепторы (TLR – Toll-lice receptors), при их стимуляции антигенами продуцируют антимикробные пептиды, ограничивая проникновение чужеродных антигенов. Однако у пациентов с АтД наблюдается снижение функции TLR. Потеря барьерной функции кожи и повышение тяжести АтД предрасполагают к микробной колонизации и хроническому воспалению кожи, включая колонизацию золотистым стафилококком и различными грибами. Было установлено, что у кератиноцитов кожи при АтД снижена способность продуцировать антимикробные пептиды, необходимые для инактивации золотистого стафилококка, грибов и предотвращения репликации вирусов [9, 10]. Интересно, что синантропные бактерии, составляющие микробиом кожи в норме, также производят антимикробные пептиды, способные контролировать рост стафилококка. Золотистые стафилококки продуцируют высокие уровни сериновых протеаз, которые могут ухудшить состояние кожного барьера [11]. Таким образом, избыточная колонизация золотистым стафилококком при АтД может ухудшать барьерную функцию кожи.
Имеются убедительные аргументы, согласно которым патогенетической основой некоторых форм АтД могут быть иммунные процессы, подавляющие терминальную дифференцировку кератиноцитов, что приводит к вторичному дефекту кожного барьера [12]:
- мутации гена FLG отсутствуют у большинства пациентов с АтД;
- у большинства детей с АтД с возра-стом наблюдается редукция симптомов болезни;
- в отличие от ихтиоза, при котором кожные симптомы проявляются с момента рождения, дебют АтД независимо от наличия или отсутствия мутации гена филаггрина наблюдается в более поздний период, но не при рождении;
- для кожи при АтД характерен широкий диапазон аномалий помимо филаггрина (лорикрин, involcucrin, corneodesmosin, клаудин и т.д.), что предполагает возможность наличия реактивных изменений дифференциации/ороговения эпидермиса;
- непосредственное воздействие на кератиноциты ИЛ-4, ИЛ-13, ИЛ-22, ИЛ-25 и ИЛ-31 подавляет экспрессию филаггрина и увеличивает функцию калликреина, который может непосредственно вызывать дисфункцию кожного барьера. ИЛ-22 непосредственно индуцирует гиперплазию кератиноцитов и подавляет экспрессию филаггрина;
- экспрессия филаггрина восстанавливается на фоне применения противовоспалительных препаратов (ингибиторов кальциневрина или кортикостероидов);
- редукция клинической активности у больных тяжелым АтД на фоне применения иммунодепрессантов, включая циклоспорин, и таргетной иммунной терапии (моноклональные антитела к ИЛ-4), что сочетается с купированием нарушений барьерных функций кожи.
Повреждения кожи при АтД всегда в той или иной степени связаны с имеющейся иммунной активацией.
При хроническом АтД неизменно присутствуют:
- увеличение инфильтрации кожи Т-клетками (примерно 10-кратное увеличение по сравнению с фоновым уровнем Т-клеток в нормальной коже);
- увеличение инфильтрации кожи миелоидными (CD11c+) дендритными клетками (также 10-кратное увеличение по сравнению с обычным уровнем кожи), при этом большинство дендритных клеток имеет «воспалительный фенотип» (BDCA1-/CD11c+) [13];
- увеличение производства цитокинов и хемокинов активированными Т-клетками и дендритными клетками при обострении АтД;
- реактивная гиперплазия эпидер-миса [14].
Тяжесть заболевания АтД связана с выраженностью и полярностью иммунной активации, а также влиянием иммунных цитокинов на эпидермальный ответ:
- индукция эпидермальной гиперплазии (ИЛ-22);
- индукция спонгиоза – межклеточного отека (ИЛ-4, ИЛ-13, ФНО – фактор некроза опухоли);
- ингибирование терминальной дифференцировки кератиноцитов (ИЛ-4, ИЛ-13, ИЛ-25, ИЛ-31, ИЛ-22, ФНО) с потенциалом обратной связи в отношении гиперплазии;
- подавление синтеза антимикробных пептидов (ИЛ-4, ИЛ-13, ИЛ-33);
- ингибирование синтеза липидов (ИЛ-4, ИЛ-13, ИЛ-31, ФНО);
- увеличение экспрессии S100A7, -8, -9 (ИЛ-22, ИЛ-17);
- индуцирование продукции TSLP (Thymic stromal lymphopoietin) кератиноидами (ИЛ-4, ИЛ-13, ФНО);
- возникновение и усиление зуда (ИЛ-31, TSLP);
- содействие антивирусному ответу (интерфероны-γ, -α, ИЛ-29).
У пациентов с АтД, имеющих повышенные уровни иммуноглобулина E (IgЕ), наблюдается селективное распространение Th2-клеток в кожном периваскулярном пространстве. Обострение АтД связано с активацией Th2, Th22, а также Th17 и соответствующих цитокинов. Показано, что не только для дебюта АтД характерно доминирование в воспалительном ответе цитокинов Th2-лимфоцитов, но и при хроническом течении болезни также сохраняется доминирующий иммунный ответ Th2-типа [15, 16].
В то время как аллергены и ирританты служат спусковым крючком (инициируют) дебюта воспаления при АтД, в последующем на первый план выходят процессы, связанные с микробной колонизацией и механизмами врожденного иммунитета.
Хронический воспалительный процесс в коже при АтД включает следующий перечень клеток врожденного и адаптивного иммунитета, функционирующих взаимосвязанно:
- различные типы дендритных клеток,
- различные подтипы Т-клеток,
- тучные клетки,
- эозинофилы,
- иные клетки.
Клинические фенотипы болезни
АтД в первую очередь определяется клиническими критериями [17]. Однако в настоящее время распространяется мнение, будто АтД является сложным синдромом с несколькими фенотипами и подтипами (эндотипами), которые клинически можно выделить по возрасту дебюта заболевания, его тяжести, ответу на терапию и триггеры (включая инфекции, аллергены, стресс).
Примерно у 80% больных АтД повышен уровень IgE в сыворотке крови, но 20% не имеют специфических IgE к пище или ингаляционным аллергенам. Тем не менее не исключено, что такие «неатопические» пациенты могут иметь IgE или аутореактивные Т-клетки к аутоаллергенам или микробным антигенам, которые обычно не измеряются [18].
Существуют и другие фенотипы АтД, в т.ч. характеризующиеся склонностью к инфицированию кожи золотистым стафилококком или развитием герпетиформной экземы. Хотя до 90% пациентов с АтД, возможно, имеют колонизацию кожи золотистым стафилококком, фактические кожные инфекции, требующие системного лечения антибиотиками, имеют немногие пациенты. Среди пациентов с АтД лишь менее 3% предрасположены к герпетиформной экземе.
Различные фенотипы возникают на основе сложного сочетания мутаций и эпигенетических эффектов на гены, контролирующие экспрессию белков в кожном барьере, врожденном и адаптивном иммунном ответе, контролирующем воздействия окружающей среды.
Определение подтипов болезни
Важность определения подтипов (эндотипов) при АтД заключается в том, что они могут быть использованы для разработки лекарственных средств прицельной (таргетной) терапии, основанной на идентификации патогенетических механизмов у конкретных пациентов.
АтД может быть разделен по генотипу и биомаркерам, отражающим иммунную поляризацию (в дополнение к клиническому фенотипу). Так, например, тяжесть болезни может быть связана с экспрессией гена FLG и его мутациями. Эти пациенты могут также служить в качестве мишени для филаггрин-ассоциированной терапии.
Примерно 80% больных имеют повышенные уровни IgE в сыворотке, часто с увеличением эозинофилии и сывороточных уровней хемокинов тимуса. Следует отметить, однако, что исследования пациентов с АтД, у которых не было выявлено специфических IgE к ингаляционным и пищевым аллергенам, продемонстрировали, что некоторые пациенты имели сывороточные специфические IgE к аутоантигенам кожи и антигенам из бактерий и грибков, колонизирующих кожу. Поэтому оправданно наличие более широкого диапазона IgE-панелей на различные экзогенные и эндогенные антигены для определения потенциальных триггеров АтД. Это может иметь значение для диагностики механизмов, вызывающих аллергическое воспаление кожи [19].
Механизмы, лежащие в основе колонизации кожи золотистым стафилококком и распространения вирусных инфекций при АтД, остаются активной областью исследования. У таких пациентов, как правило, очень выражены проявления атопии с повышенным уровнем сывороточного IgE и эозинофилией периферической крови. Это отражает патогенетические механизмы, связанные с высоким уровнем активации Th2 и продукцией данными клетками цитокинов, что, как известно, способно уменьшать барьерную функцию кожи, повышать колонизацию кожи золотистым стафилококком, уменьшать производство антимикробного пептида (AMP) и ухудшать качество врожденных иммунных ответов.
В то же время, поскольку герпетиформная экзема является крайне редкой, а инфицированность вирусом простого герпеса (ВПГ) очень частой, вероятно, что дополнительные иммунологические и генетические факторы способствуют возникновению AтД, связанного с герпетиформной экземой (ADEH+). Недавно было обнаружено, что такие пациенты хорошо реагируют на лечение интерфероном-α2b. По-видимому, тяжесть заболевания связана с величиной и полярностью иммунной активации, а также влиянием эффектов иммунных цитокинов на эпидермальный ответ.
Определение биомаркеров при АтД позволит определить пациентов, имеющих различные варианты иммуногенеза болезни. В дальнейшем результаты эпидермальной протеомики, геномики, определения биомаркеров крови в сочетании с клиническим фенотипом предложат бóльшую точность в определении эндотипов АтД [20].
Современные подходы к терапии
Таким образом, в основе патогенеза АтД лежит сочетание дисфункции кожного барьера и иммунного воспаления кожи. В связи с этим основными компонентами в терапии являются подходы, направленные на репарацию эпидермального барьера, восстановление микробиома, восстановление гидролипидной мантии в сочетании с активной противовоспалительной терапией, включая топические глюкокортикостероиды, ингибиторы кальциневрина, контроль инфекционных осложнений, элимнацию триггеров и этиологических факторов, в т.ч. аллергены, раздражители и эмоциональные триггеры, таргетную терапию, направленную на купирование конкретных патогенетических процессов. В основе современной терапии этой болезни лежит поэтапный подход, который зависит от тяжести заболевания, фазы болезни, площади поражения кожи [21].
В настоящее время в терапию АтД внедряется метод таргетированного лечения (введение целевых точек), его осмысление, формирование конкретных мишеней. Основой для внедрения такого подхода послужила дешифровка патогенеза заболевания [22].
В лечении пациентов с хроническим течением АтД важно учитывать имеющееся снижение содержания белков эпидермального барьера и повышение трансэпидермальной потери воды, даже в период ремиссии. В связи с этим важно поддерживать эпидермальный барьер, применяя смягчающие средства топической терапии даже в периоды ремиссии.
Пациентами, которые не отвечают на обычные подходы к лечению, может быть использован ряд альтернативных стратегий, в т.ч. циклоспорин, метотрексат, азатиоприн, ИЛ-6-блокада, ультрафиолетовое облучение. Недавние исследования прицельной (таргетной) терапии с участием молекулярных и клеточных биомаркеров показывают, что фенотип хронического АтД может быть курабельным при использовании таких методов лечения.
Тяжелый АтД может быть связан с системной иммунной активацией, что объясняет частую потребность в использовании системных препаратов для иммунного подавления, недостаточность топической терапии кожи, вовлеченной в патологический процесс. В терапии этих пациентов необходимо добиваться применения методов топической терапии, распространяющейся на весь кожный покров.
Существенное расширение наших знаний о молекулярных механизмах, лежащих в основе развития этой болезни, позволяет сформулировать и выявить потенциальные цели и подходы при лечении.
К ним относятся:
- новые способы сокращения трансэпидермальной потери влаги и повреждения корнеоцитов;
- новые способы воздействия на установленные молекулярные мишени (такие, как рецепторы гистамина, интерлейкинов и других цитокинов Т-клеток);
- выявление новых молекулярных мишеней (например, Toll-подобные рецепторы; белки плотного контакта – tight junction proteins).
Хорошо зарекомендовавшие себя варианты лечения, такие как смягчающие вещества, кортикостероиды, топические ингибиторы кальциневрина, сохраняют свои позиции в лечении АтД. Среди новых подходов, которые могут присоединиться к ним в ближайшем будущем, являются сфинганин (предшественник церамидов 1 и 3), каннабиноиды, моноклональные антитела.
Барьерная функция и ее коррекция
Сложный процесс эпидермальной дифференцировки нарушается при АтД, что представляет собой множество потенциальных мишеней для терапевтического вмешательства. Корнеоциты прикреплены друг к другу с помощью corneodesmosomes. Имеется особый фермент – SCCE (Stratum Corneum Chymotryptic Enzyme), способный расщеплять corneodesmosomes. Он становится активным в верхних слоях рогового слоя после того, как нарушается функция его антагониста – ингибитора протеазы LEKTI (Lymphoepithelial Kazal-Type-Related Inhibitor). У пациентов с АтД наблюдается повреждение corneodesmosomes, нарушается сцепление клеток, повышается их отшелушивание. Барьерная дисфункция кожи при АтД характеризуется повышенной трансэпидермальной потерей воды, снижением гидратации рогового слоя. В норме роговой слой кожи имеет слабокислый рН вследствие естественного увлажняющего фактора NMF (Natural Moisturizing Factor), который состоит из продуктов деградации филаггрина молочной и уроканиновой кислот (UCA – Urocanic Acid). Тогда как оптимальное значение рН для SCCE находится в щелочной области, слегка кислый рН рогового слоя защищает кожу от деградации. Использование щелочного мыла повышает рН рогового слоя, что увеличивает осыпание корнеоцитов. Это служит научной основой для рекомендаций пациентам с АтД заменять классическое мыло на специальные моющие средства.
Добавление компонентов филаггрина в увлажняющие агенты является многообещающей стратегией лечения. На основании клинических данных хорошо известно, что малые молекулы, мочевина и глицерин улучшают барьерную функцию кожи. Добавление вазелина к барьерным стабилизирующим кремам имеет эффект уплотнения и уменьшает трансэпидермальную потерю воды. Также керамиды являются важным компонентом двойного липидного слоя, входящего в гидролипидную мантию кожи, при этом их содержание обратно коррелирует с потерей трансэпидермальной воды. В связи с этим добавка церамидов в увлажняющие кремы может быть важным терапевтическим шагом.
Добавление сфинганина, который метаболизируется до церамидов 1 и 3, также считается перспективной терапевтической стратегией.
Препараты, направленные на нарушенную функцию барьера на уровне белков плотного контакта, как представляется, остаются также перспективным средством для лечения АтД.
Иммунные мишени терапии
Для прицельной (таргетной) терапии рассматриваются препараты, ориентированные на многие мишени иммунного ответа при АтД, включая ИЛ-4, TSLP, Th22, Th17. Выбор иммунных таргетных терапевтических средств для пациентов с различными степенями тяжести заболевания или признанных его фенотипов не может быть случайным, он должен ориентироваться на степень активации различных иммунопатологических механизмов, как системных, так и органоспецифических (кожа).
В настоящее время исследуется эффективность биологических препаратов, воздействующих на различные точки иммуногенеза АтД: блокаторы ФНО, антитела к IgE (омализумаб), антитела к ИЛ-5 (меполизумаб). Имеются публикации об отдельных наблюдениях по применению при АтД анти-CD20 (ритуксимаб), анти-ИЛ-12/23 (устекинумаб), анти-ИЛ-6R (тоцилизумаб). Продолжающиеся исследования анти-ИЛ-4R, -ИЛ-13, -ИЛ-22, -ИЛ-31, -TSLP, -CRTH2 (Chemoattractant Receptor-homologous molecule).
Среди потенциальных мишеней лечения АтД ранее рассматривался ИЛ-5. В качестве соответствующего лекарственного средства – антитела к ИЛ-5 (меполизумаб). На фоне терапии данным препаратом было отмечено улучшение отдельных параметров экземы. Однако в данном исследовании не было получено реализации поставленной цели – быстрого и значительного клинического улучшения при лечении тяжелых вариантов АтД и снижения содержания тимус-ассоциированного регуляторного хемокина (Th2-хемокина) TARC, производимого кератоцитами в пораженной аллергическим воспалением коже и контролирующего накопление в ней клеток, участвующих в воспалении. Терапия меполизумабом не предотвращала экзематозную реакцию в патч-тестах. Приток эозинофилов в ткань кожи при проведении патч-теста сокращался незначительно, хотя наблюдалось сокращение числа эозинофилов в периферической крови [23].
Большие надежды возлагались также на применение анти-IgE-антител – препарата омализумаб, хорошо зарекомендовавшего себя при лечении атопической БА. Первоначальные наблюдения за пациентами с сочетанием БА и АтД продемонстрировали определенные успехи при применении омализумаба в купировании кожных проявлений [24]. Последующее рандомизированное проспективное двойное слепое плацебо-контролируемое исследование не продемонстрировало значимого клинического эффекта омализумаба в лечении АтД [25].
В то же время имеется подгруппа пациентов с АтД, которым терапия омализумабом может быть показана и эффективна: к ним относятся пациенты с преобладанием в иммуногенезе IgE-опосредуемых аллергических реакций.
В настоящее время имеются отдельные клинические наблюдения других биологических препаратов. Использование ритуксимаба (направлен против В-клеточного антигена CD20), первоначально разработанного для B-клеточной лимфомы, а в дальнейшем продемонстрировавшего эффективность при лечении ревматоидного артрита и других аутоиммунных заболеваний, у шести больных АтД продемонстрировало значительное снижение тяжести заболевания [26].
Биологические препараты, утвержденные для лечения псориаза, мишенью которых является ФНО, были оценены пациентами с АтД, но не показали многообещающего успеха. Напротив, у некоторых пациентов было даже отмечено значительное ухудшение течения болезни [27].
ИЛ-23- и ИЛ-12-блокирующие антитела – препарат устекинумаб (антитела к субъединице р40 этих интерлейкинов), так же как и блокаторы ФНО, уже одобрены для лечения псориаза. При лечении пациентов с сочетанием псориаза и АтД была отмечена редукция симптомов последнего. В дальнейшем получено значительное улучшение течения АтД при лечении устекинумабом пациента с тяжелой формой болезни (без сочетания с псориазом) уже после 4 недель терапии [28]. Данные результаты обнадеживают, но необходимы дальнейшие исследования.
Сообщается о небольшой группе из 3 пациентов, в терапии которых применяли антагонист рецептора ИЛ-6 тоцилизумаб; у каждого пациента отмечен хороший успех терапии. Однако, в связи с тем что ИЛ-6 является очень важным цитокином для иммунной защиты против бактериальных инфекций, в этой группе пациентов были отмечены случаи инфекционных осложнений в виде бактериального конъюнктивита и бактериального бурсита, что может ограничить его дальнейшее использование [29].
Исследования таргетной биологической терапии АтД в настоящее время продолжаются. Ранее было отмечено, что при АтД имеет место доминирование Th2-цитокина ИЛ-4, что легло в основу соответствующей терапевтической стратегии. Эти подходы не имели успеха, однако в настоящее время продолжают активно изучаться, прежде всего применение при АтД дупилумаба – антитела к рецепторам IL-4 и IL-13. Дупилумаб продемонстрировал положительные эффекты при АтД в контролируемых исследованиях – сочетание применения этих антител с местными стероидами привело к убедительным результатам [30]. Это подтверждает не только центральную роль Th2-цитокинов ИЛ-4 и ИЛ-13 для АтД, но и то, что эти цитокины значимы не только на ранней стадии воспаления, но и в хронической персистирующей фазе воспаления. ИЛ-4 Rα-антагонист (дупилумаб) приводит к значительному клиническому улучшению пациентов со среднетяжелым и тяжелым течением АтД. Снижение гиперплазии при 4-недельном применении дупилумаба было существенно выше, чем терапия циклоспорином в дозе 5 мг/кг курсом 12 недель пациентов с аналогичной активностью болезни [31].
Аналогичным дупилумабу (вышеуказанные антитела против ИЛ-4 Rα и ИЛ-13Rα) эффектом может обладать также питракинра (Aerovant) – белок, связывающий рецептор α ИЛ-4, являющийся также антагонистом эффектов ИЛ-4 и ИЛ-13. ИЛ-13 является цитокином Th2-лимфоцитов, служит мишенью и лебрикизумаб, моноклональных антител, планирующихся к исследованию при АтД.
Изучаются антитела, мишенью которых является ИЛ-31. Цитокин ИЛ-31 связан с Th2-ассоциирвоанным воспалительным ответом и, как представляется, в частности, ответствен за формирование зуда при АтД.
В настоящее время проводится изучение при АтД моноклональных антител, направленных против ИЛ-22, Т-клеточного цитокина, относящегося к семейству Th17, но в отличие от ИЛ-17 при АтД значительно усиливающего свою активность в пораженной коже (https://clinicaltrials.gov).
Другие экспериментальные подходы к лечению АтД направлены на CRTH2: известный как маркер Th2-клеток, он является рецептором для простагландина D2 (PGD 2), высвободившегося из тучных клеток. Блокирование CRTH2 служит концепцией лечения АтД, первые результаты многообещающи.
TSLP – цитокин семейства ИЛ-7, является важным медиатором Th2-лимфоцитов. Он высвобождается кератиноцитами под влиянием, в частности, дендритных клеток, принимая опосредованное участие в формировании зуда при АтД [32]. В настоящее время проводятся исследования, в которых в качестве потенциальной мишени при лечении АтД рассматривается TSLP [33].
Заключение
АтД – наиболее распространенное хроническое воспалительное заболевание кожи у детей [34]. Он часто ассоциируется с формированием пищевой аллергии, а в последующем и респираторной аллергии, включая БА и АР, что имеет образное название «атопический марш».
Много методов уже было описано для лечения АтД, но ни один из них не является исцеляющим. Сочетание смягчающей терапии, противовоспалительной терапии и терапии, влияющей на микробиом кожи, кажется оптимальной для большинства пациентов. Последние результаты исследования патогенеза АтД позволили выявить отклонения в дифференцировке эпидермального эпителия, приводящего к дефектам рогового слоя кожи. Это служит потенциальной основой трансдермального проникновения аллергенов и системной IgE-опосредованной сенсибилизации. Кожа пациентов с АтД предрасположена также к колонизации или инфицированию патогенами, в первую очередь стафилококками, грибами, вирусами группы герпеса. Причины данных патологических изменений со стороны кожного барьера сложны и обусловлены комбинацией генетических, экологических и иммунологических факторов. Эти факторы, вероятно, обусловливают неоднородность дебюта АтД, тяжести и течения этой болезни.
Недавние исследования показывают, что предупреждение формирования АтД может быть достигнуто путем ранних вмешательств, обеспечивающих сохранность и эффективность кожного барьера. Детям со стартовавшим АтД необходимо обеспечить терапию, которая позволит эффективно контролировать как местные, так и системные проявления иммунной активации. Раннее оптимальное терапевтическое вмешательство может улучшить долгосрочные результаты лечения АтД, уменьшить системную сенсибилизацию аллергенами, которая приводит к формированию коморбидных аллергических заболеваний дыхательных путей и патологии желудочно-кишечного тракта.


