
Введение
В настоящее время наблюдается значительное увеличение числа опухолевых заболеваний, которые выступают в качестве одной из главных причин потери трудоспособности и смертности во всем мире. В связи с этим выяснение механизмов канцерогенеза является одной из наиболее важных современных медико-биологических проблем. В последние годы были получены результаты, позволяющие пересмотреть роль гипоксии в образовании сóлидных опухолей. Превращение нормальных клеток в канцерогенные характеризуется не только их морфологическими изменениями, но и перестройкой функциональной активности, а также глубокими изменениями генетического аппарата и метаболизма. Все эти изменения сопровождаются приобретением новых признаков, свойственных опухолевым клеткам. Они перестают отвечать на воздействие различных регуляторных систем, ответственных за согласованную активность клеток в многоклеточном организме; подвергаются многократным мутациям генов на разных этапах существования; характеризуются нестабильностью генома; становятся автономными; делятся неограниченным образом; не дифференцируются; становятся устойчивыми к действию иммунокомпетентных клеток; не удаляются из организма с помощью апоптоза; не подвергаются контактному торможению; становятся инвазивными; образуют метастазы; существуют в условиях длительной ишемии; активируют опухолевый ангиогенез; перестраивают метаболизм, в т.ч. энергетический обмен.
Превращение нормальной клетки в трансформированную – процесс многостадийный. При этом выделяют несколько важных стадий онкогенеза:
- Возникновение опухолевой клетки вследствие мутаций ряда генов, прежде всего генов, контролирующих клеточное деление, – стадия инициации.
- Активация опухолевой клетки в результате блокады антиопухолевых механизмов, что приводит к делению трансформированной клетки и образованию клона идентичных дочерних клеток, – стадия промоции.
- Стадия опухолевого прогрессирования, когда в результате дополнительных мутаций из клеток первичного клона образуются новые клоны клеток, отличающихся по своим свойствам от исходных клеток.
Стадия инициации связана с изменением структуры ДНК и образованием трансформированных клеток. Стадия промоции характеризуется возникновением дополнительных мутаций, нарушением механизмов репарации ДНК в трансформированной клетке, подавлением антиканцерогенной активности, стимуляцией деления такой клетки с участием опухоль-инициирующих факторов и формированием клона канцерогенных клеток из одной материнской клетки. Между первой и второй стадиями могут проходить годы. Третья стадия – это процесс пролиферации видоизмененных клеток первичного клона, подвергшихся дополнительным изменениям генетического аппарата, из которых образуется множество клонов опухолевых клеток, отличающихся по своим свойствам; естественный отбор наиболее жизнеспособных клонов и образование опухоли из таких клонов; малигнизация опухолевых клеток, приобретение способности к инвазии и метастазированию [1].
На этапе промоции трансформированная опухолевая клетка начинает делиться и превращается в недифференцированные, молодые клетки одного клона, которые формируют первоначальный агрегат клеток.
В основе подобной пролиферации лежат разные причины, включая активацию онкогенов или подавление активности антионкогенов, блокаду репарации ДНК и другие регуляторные процессы [2]. Следует особо отметить, что опухолевые клетки не распознаются и не удаляются из организма иммунной системой, при этом ингибируются внеклеточные и внутриклеточные механизмы запуска апоптоза, а также блокируются инактивация и удаление видоизмененных собственных клеток, не выполняющих своей функции. Активация деления трансформированных клеток часто бывает связананой с дополнительными множественными мутациями соответствующих генов и нестабильностью генома трансформированной клетки. Все это способствует развитию этапа прогрессирования опухоли. Новые видоизмененные клоны опухолевых клеток отличаются друг от друга и подвергаются естественному отбору по разным признакам, что способствует выживанию наиболее жизнестойких, приспособленных к конкретным условиям опухолевых клеток с высоким пролиферативным потенциалом, которые формируют макро-опухоль. На этом этапе происходит малигнизация опухолевых клеток и их инвазия в окружающие ткани, а также метастазирование, ответственное за распространение клеток злокачественной опухоли по организму [3, 4].
Мутагены
Превращение нормальной клетки в опухолевую (трансформация клетки) является критическим этапом онкогенеза. Трансформация клетки на этапе инициации и последующих этапах, связанная с изменениями структуры генов, является результатом воздействия мутаций или переноса с помощью вирусов генов человека, контролирующих деление клеток (онкогенов). Мутации, лежащие в основе инициации, возникают в результате воздействия разных причин, включая химические, физические и биологические факторы – мутагены. К химическим мутагенам относятся разнообразные соединения внешней среды – экзогенные мутагены и эндогенные агенты, которые, как и другие мутагенные факторы, влияют прежде всего на собственные неактивные онкогены (прото-онкогены) в геноме человека и переводят их в форму онкогенов. Физические канцерогены, как и химические, весьма разнообразны. Они представлены ионизацирующей радиацией, ультрафиолетовым и ультразвуковым излучениями, механическими и температурными внешними воздействиями. В последние годы особое внимание уделяют радикалам кислорода, которые образуются в клетках при оксидативном стрессе и генерации больших количеств радикалов NO, например, под влиянием индуцибельной формы NO синтазы – iNOS. Такие радикалы генерируются в большом количестве в непосредственной близости от ДНК ядра и кольцевых ДНК митохондрий, они также способны вызывать мутации генов [5].
Многие биологические факторы, инициирующие деление клеток, являются продуктами онкогенов (онкобелками). К таким онкобелкам относятся различные факторы роста клеток: фактор роста эндотелия сосудов (VEGF – Vascular Endothelial Growth Factor), фактор роста тромбоцитов (PDGF – Platelet-derived growth factor, и др). Неадекватное образование факторов роста, а также изменение активности их рецепторов играют важную роль в последующем запуске деления трансформированных клеток и формировании опухолей.
Изменения биологических свойств опухолевых клеток при гипоксии
Опухолевые и высокоагрессивные клетки, быстро растущие сóлидные опухоли начинают подвергаться воздействию гипоксии с самых ранних этапов онкогенеза, начиная с этапа промоции (первичные опухоли 2–3 мм3) и заканчивая опухолевым прогрессированием [3, 4]. Гипоксия опухолей является следствием двух процессов – неадекватно низкого кровоснабжения и высокой скорости использования кислорода при активном делении трансформированных клеток. Важно отметить, что даже после неоваскуляризации опухоли доставка кислорода продолжает отставать от количества кислорода, необходимого для быстрого деления клеток, в связи с чем гипоксия остается характерным признаком опухолевого роста [6].
В течение длительного времени считалось, что гипоксия ответственна исключительно за гибель опухолевых клеток, а восстановление поступления кислорода и его использование для синтеза АТФ в митохондриях способствует увеличению скорости деления канцерогенных клеток, росту опухолей и сопровождается неблагоприятным исходом заболевания. Однако постепенно накапливались результаты, которые свидетельствовали о том, что гипоксия является ключевым рычагом, способствующим делению опухолевых клеток, их выживанию, а с дефицитом кислорода связано не только образование областей некроза в опухолях, но и существенные изменения биологических свойств опухолевых клеток [7].
Гипоксия ответственна за адаптацию и выживание канцерогенных клеток в неблагоприятных условиях. Под влиянием гипоксии происходит естественный отбор жизнеспособных мутантных клеток, деление которых сопровождается образованием различных клонов, из которых в конечном итоге формируется злокачественная опухоль. Большинство механизмов, активируемых гипоксией, участвуют в опухолевом прогрессировании, а одно из следствий ее влияния на клетки связано с их метастазированием.
В результате этих процессов при гипоксии развивается устойчивость к апоптозу и резистентность по отношению к разным видам лечебных воздействий. Таким образом, гипоксия выступает в качестве движущей силы опухолевого прогрессирования [7, 8].
Среди генов, активированных при гипоксии, значительный интерес представляют гены, участвующие в опухолевом ангиогенезе. Снижение активности таких генов тормозит рост опухоли [9]. Однако гипоксия не только стимулирует образование сосудов в опухолях, но и влияет на взаимодействие между опухолевыми клетками, а также на их связь с внеклеточным матриксом. Так, установлено, что гипоксия подавляет экспрессию Е-кадгерина в эндотелиальных клетках микрососудов [10, 11]. Потеря кадгерина, участвующего в межклеточных контактах, рассматривается как начальный этап в опухолевом прогрессировании. Нарушение межклеточного взаимодействия и потеря контактного торможения деления канцерогенных клеток играют также важную роль в их последующем метастазировании. Гипоксия опухоли ответственна за селективный отбор мутантных генов и канцерогенных клеток, устойчивых к развитию апоптоза [12, 13].
Установлено, что повторные эпизоды кратковременной гипоксии приводят к предпочтительному отбору мутаций гена белка р53, что делает этот белок неактивным и подавляет передачу апоптозного сигнала в цитоплазме клеток на эффекторные каспазы.
В результате опухолевые клетки с такой мутацией становятся резистентными к апоптозу, индуцированному гипоксией [11], а также устойчивыми к разным видам лечения, включая лучевую терапию [7].
Внутриклеточные сигнальные процессы, участвующие в канцерогенезе
Гипоксия стимулирует несколько внутриклеточных сигнальных путей, передающих информацию на факторы транскрипции генов, которые поступают в ядро и стимулируют различные гены, в т.ч. онкогены. Продукты таких генов изменяют метаболизм трансформированных клеток, способствуют их озлокачествлению и диссеминации по всему организму. Установлено, что в этих процессах важную роль играют члены семейства митоген-активированных протеинкиназ (МАРК – mitogen-activated protein kinase) [14–16].
Суперсемейство митоген-активированных протеин киназ (МАРКs) состоит из трех основных протеин-киназных семейств:
- Протеин киназ, регулируемых внеклеточными сигналами (ERKs – extracellular signal-regulated kinases)).
- c-Jun N-теминальных киназ (JNKs – c-Jun N-terminal kinases).
- Семейство киназ р38.
Каждое из указанных семейств МАРКs играет важную роль в регуляции клеточного метаболизма, интеграции внутриклеточных сигнальных путей, в экспрессии определенных групп генов. МАРКs контролируют рост, деление, дифференцировку, механизм гибели клеток, в т.ч. апоптоз и его блокаду, а также ответы клеток на разнообразные воздействия, включая температурные, осмотические и оксидативные виды стресса. Установлено, что МАРКs играют одну из ведущих ролей в адаптации клеток к внешним неблагоприятным воздействиям, в т.ч. гипоксии и аноксии [17]. Между внутриклеточными путями МАРКs, контролируемыми разными семействами протеин киназ, существует тесная взаимосвязь: протеин киназы семейства р38, активируемые при стрессах и цитокинами воспаления, в конечном итоге не только влияют на транскрипцию генов, контролирующих ремоделирование хроматина и цитоскелета клеток, но и по перекрестным путям индуцируют активность протоонкогенов с-fos, активацию ATF-2 и c-Jun киназы. Большое значение в активации генов опухолевых клеток при гипоксии имеет внутриклеточный сигнальный каскад, связанный c фосфатидилинозитол 3-киназой (PI3K) и Akt протеин киназой (или протеин киназой В-PKB) [18].
Роль фактора транскрипции генов, активируемого гипоксией, в биологической активности опухолевых клеток
В течение длительного времени природа сенсора гипоксии в эукариотических клетках оставалась неизвестной. В работах последних лет было установлено, что в качестве такого сенсора выступает фактор, индуцируемый гипоксией (HIF – hypoxia-inducible factor). Этот фактор является ядерным рецептором, который активирует транскрипцию более 200 генов, в т.ч. генов, участвующих в опухолевой трансформации и образовании опухолей. Однако продукты таких генов играют критическую роль не только в канцерогенезе, но и в устойчивости к противоопухолевой терапии, в т.ч. к химио- и радиотерапии в условиях гипоксии. Они влияют на энергетический обмен опухолевых клеток (метаболизм глюкозы); адаптацию к гипоксии и их выживание; инвазию и метастазирование; ангиогенез и усиление прорастания мелких кровеносных сосудов непосредственно в опухоль [19]. Гипоксия опухоли увеличивает экспрессию HIF в канцерогенных клетках, стимулирует онкогены, увеличивает скорость роста и деления этих клеток. Доказано, что высокий уровень этого фактора выступает в качестве неблагоприятного индикатора течения болезни и связан с увеличением смертности пациентов при опухолях разных видов, в т.ч. молочной железы, желудка, эндометрия и яичников [36]. Таким образом, гипоксия участвует в локальном и системном прогрессировании опухолей, а также вносит свой вклад в резистентность к радио- и химиотерапии, а HIF является важнейшим регулятором транскрипции различных генов опухолевых клеток.
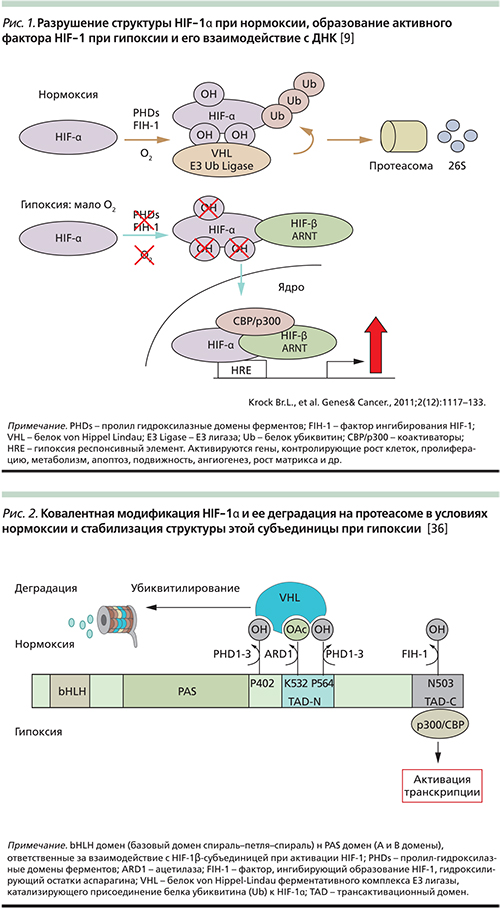
Фактор транскрипции генов (HIF-1) в активном состоянии представляет гетеродимер, состоящий из двух типов субъединиц, каждая из которых является базисным транскрипционным фактором: кислород-зависимой, индуцибельной HIF-1α субъединицей и конститутивно экспрессируемой HIF-1β (ARNT) субъединицей, ответственной за связывание с ДНК. Регуляция стабильности и активность HIF-1α зависят от наличия кислорода. При нормальном содержании кислорода HIF-1α-субъединица подвергается ковалентным модификациям структуры, сперва гидроксилированию с участием ферментов пролил гидроксилаз (PHDs – Prolyl Hydroxylase Domain isoforms), которые присоединяет гидроксильную группу к остаткам пролила в α-субъединице HIF, а затем происходит взаимодействие с белком VHL (von Hippel-Lindau). Образовавшийся комплекс действует на фермент лигазу, который катализирует многократное присоединение к этой субъединице низкомолекулярного белка убиквитина (рис. 1). Белок VHL является компонентом лигазы, который распознает гидроксилированную форму HIF-1α. Модифицированная таким образом HIF-1α-субъединица поступает на протеасому и разрушается (рис. 2).
Установлено, что при гипоксии активность кислород-зависимых пролил гидроксилаз (PHDs) падает, что увеличивает уровень HIF-1α-субъединицы и ее стабильность. В результате образуются функционально активные гетеродимеры HIF-1, состоящие из HIF-1α/HIF-1β (ARNT – aryl hydrocarbon receptor nuclear translocator), которые связываются с ДНК и взаимодействуют с коактиватором р300/СВР, регулирующим транскрипцию различных генов [20]. Важно отметить, что в результате действия HIF-1 на ДНК изменяется экспрессия группы генов, в т.ч. ответственных за процессы адаптации и увеличения жизнеспособности опухолевых клеток (например, возрастает образование VEGF, который участвует в опухолевом ангиогенезе).
Помимо этого установлено, что с гипоксией и активацией HIF-1 связана экспрессия генов – переносчиков глюкозы в клетки, а также образование всех ферментов гликолиза в цитоплазме опухолевых клеток разного вида, ответственных за переключение энергетического обмена в опухолевых клетках с окислительного фосфорилирования в митохондриях на гликолиз в цитоплазме. Феномен такого переключения был впервые описан О. Варбургом в 1920-е гг. и получил названия фенотипа Варбурга, характерного для опухолевых клеток [21].
Cнижение активности HIF-1 как новый подход к подавлению роста и метастазирования опухолей
В условиях гипоксии в опухолевых клетках глубокие изменения претерпевают сигнальные пути, регулирующе метаболизм, энергетический обмен, пролиферацию, ангиогенез и клеточную гибель. Выраженная адаптация к гипоксии канцерогенных клеток позволяет им не только выживать и активно расти и делиться, но и проявлять резистентность к терапии. В связи с этим сигнальные пути гипоксии и их мишени выступают в качестве многообещающих новых мишеней для терапевтических воздействий на канцерогенез [7].
Как уже отмечалось, регуляция и активность фактора HIF-1 играет одну из ведущих ролей в жизнедеятельности канцерогенных клеток. В ряде экспериментальных исследований показано, что снижение или подавление активности HIF-1 в опухолевых клетках тормозит их рост и метастазирование. На основании таких результатов было предположено, что воздействие на разные сигнальные пути, а также активность ключевых ферментов, регулирующих HIF-1α и HIF-1, с помощью «антагонистов» HIF-1 позволяет создавать новый тип антиопухолевых лекарственных препаратов [37].
Один из таких подходов основан на блокаде гипоксия-отвечающих элементов (HRE – hypoxia-response element), участвующих во взаимодействии HIF-1 c ДНК. Так, в работе Y. Hasebe и соавт. в результате скрининга был идентифицирован ряд флавоноидов и гомоизофлавоноидов, ингибирующих активацию HRE в условиях гипоксии [22]. Одним из таких активных соединений был метил-офиопогонанон В (МОВ – methyl ophiopogonanone В), в концентрациях 3–9 мкг/мл подавляввший активность HRE. При гипоксии в более высоких дозах МОВ (10–20 мкг/мл) блокировал экспрессию мРНК фактора роста эндотелия сосудов (VEGF) и снижал его уровень в опухолевых клетках линии HepG2. С помощью антител против HIF-1α (Вестерн блотинга) было показано, что МОВ вызывал дестабилизацию HIF-1α и разрушение белка этой субъединицы, что зависело от активности протеасом и супрессора опухолей р53. Миграция эндотелиальных клеток пупочной вены человека в процессе ангиогенеза также подавлялись МОВ. На основании этих результатов был сделан вывод: антиопухолевое действие МОВ связано с блокадой ангиогенеза.
В другой работе был выбран аналогичный подход для подавления активности HIF-1 [23]. С этой целью в результате скрининга 15 тыс. соединений было отобрано 3, которые эффективно блокировали связывание HIF-1 c ДНК в результате ингибирования элемента, отвечавшего на гипоксию (HRE), участвовавшего во взаимодействии HIF-1 с ДНК. Эти 3 соединения не снижали уровни белка HIF-1α в опухолевых клетках рака молочной железы линии MDA-468, но подавляли рост этих клеток, а также индукцию генов-мишеней HIF-1 при гипоксии, в т.ч. активность гена VEGF. Таким образом, антиканцерогенные эффекты этих трех соединений были обусловлены ингибированием активации генов, контролируемых HIF-1.
Показано также, что связывание ионов железа с помощью хелатных комплексонов и их удаление из пролил гидроксилаз подавляют разрушение HIF-1α-субъединицы и имитируют действие гипоксии на ее стабилизацию. Эти данные свидетельствуют о том, что ионы Fe2+ нужны для взаимодействия ферро-протеина с О2 и играют важную роль в гидроксилировании остатков пролила и последующей деградации HIF-1α [24].
Однако следует отметить, что увеличение доставки в опухолевые клетки кислорода является одним из наиболее эффективных путей естественной регуляции активности HIF-1, в связи с тем что именно наличие кислорода активирует ферменты, ответственные за разрушение HIF-1α, подавление его взаимодействия с HIF-1β и образование активного HIF-1. Таким образом, доставка кислорода в случае карцерогенеза – критический этап, блокирующий активацию HIF-1 и в результате угнетающий образование опухоли и ее метастазирование.
По этой причине целесообразно остановиться на препаратах с выраженным антигипоксантным действием. В связи с этим мы более подробно остановимся на описании свойств депротеинизирванного гемодеривата (Актовегин).
Влияние на гипоксию клеток препаратов биологического типа
Препарат Актовегин, долгое время широко применяющийся в клинической практике для лечения острой и хронической недостаточности церебрального кровообращения, заболеваний периферических вен и артерий, диабетической полиневропатии и ряда других состояний, представляет собой безбелковый гемодиализат крови молочных телят. В состав препарата входит около 200 низкомолекулярных компонентов с молекулярной массой, не превышающей 5000 Да. Благодаря низкому молекулярному весу компонентов Актовегина (пептиды, аминокислоты, инозитолфосфоолигосахариды, сфинголипиды, биогенные амины, полиамины, жирные кислоты, ацилкарнитины и многие другие) они проникают через различные барьеры, включая гематоэнцефалический, и оказывают разнообразные эффекты на клетки как центральной и периферической нервной системы, так и других органов и тканей. Препарат обладает инсулиноподобным действием (активирует систему переносчиков глюкозы через плазматические мембраны клеток и кислород-зависимый энергетический обмен в митохондриях) в условиях ишемии. В условиях гипоксии Актовегин защищает от повреждений и гибели гепатоциты, кардиомиоциты, нейроны и клетки других видов [25, 26].
Ряд авторов отметили также антиоксидантные свойства препарата. Это было продемонстрировано как на первичной культуре нейронов гиппокампа крыс [27], так и на перевиваемых нейронах человека in vitro и фагоцитах крови [28–31]. Так, в ряде наших работ Актовегин эффективно подавлял образование и секрецию радикалов кислорода фагоцитами крови, стимулированных бактериальным трипептидом FMLP (Formyl-Methionyl-Leucyl-Phenylalanine) или форболовым эфиром, которые генерировались мембранным ферментативным комплексом – НАДФН-оксидазной системой [28, 29]. Помимо этого Актовегин снижал внутриклеточный уровень радикалов кислорода, измеренный с помощью флуоресцентного красителя в цитоплазме опухолевых нейронов линии SK-N-SH, подвергшихся воздействию высокой концентрации пероксида водорода (100 мкМ) [30, 31].
Антигипоксантный эффект препарата был продемонстрирован на ряде моделей в эксперименте. Так, Актовегин как антигипоксант усиливал потребление кислорода на моделях изолированных почек и скeлетных мышц лабораторных животных [32, 33].
Механизмы, ответственные за усиление кислород-зависимого метаболизма митохондрий под влиянием гемодиализного экстракта крови телят, были изучены в работе Т. Kuninaka и соавт. [34]. Установлено, что эти эффекты зависели от нахождения митохондрий в разных функциональных состояниях по Б. Чансу, природы используемых митохондриальных субстратов и их концентраций, а также ингибирования АТФазы митохондрий и применения разобщителей окислительного фосфорилирования. На основании полученных результатов авторы пришли к заключению о том, что Актовегин, энергетические субстраты и разобщители влияют на дыхание митохондрий по разным механизмам. Препарат усиливал синтез АТФ и увеличивал потребление кислорода митохондриями, что свидетельствует о его антигипоксическом действии. Таким образом, в основе антигипоксического эффекта Актовегина лежит усиление потребления кислорода митохондриями, которое тесно связано с окислением метаболита глюкозы пирувата в матриксе этих органелл. Эти данные позволяют предположить, что Актовегин, снижая гипоксию митохондрий, увеличивая поступление кислорода в клетку и митохондрии, может потенциально усиливать разрушение субъединицы HIF-1α и предупреждать образование активированного фактора транскрипции генов – HIF-1. В результате не происходит активации HIF-1-зависимых генов, ответственных в т.ч. за экспрессию всех ферментов гликолиза, процесс ангиогенеза и развитие адаптации опухолевых клеток к неблагоприятным условиям гипоксии. Это предположение хорошо согласуется с физиологическими эффектами Актовегина, который улучшает когнитивные процессы в теменной части коры мозга, что способствует восстановлению памяти у пожилых людей. Причиной таких защитных эффектов Актовегина является его положительное действие на нейроны центральной нервной системы при гипоксическом стрессе, улучшение функциональной активности митохондрий нейронов коры мозга и гиппокампа у старых крыс [35], а также снижение генерации радикалов кислорода в этих условиях [27, 28]. В работе Hoyer и соавт. изучали влияние Актовегина на метаболизм глюкозы и энергетический обмен в коре мозга и гиппокампе старых двухлетних самцов крыс Wistar через 15 минут после полной ишемии и последующей реперфузии в течение 60 минут, 24 часов, 48, 72 и 96 часов. После полной ишемии определяли изменения обмена энергетических субстратов путем измерения концентрации глюкозы, лактата, креатин фосфата (CrP) и АТФ, которые первоначально быстро восстанавливались до нормальных значений. Однако уже через 24 часа и в более выраженной форме спустя 48 и 72 часа после начала реперфузии было зарегистрировано нарушение баланса энергетических субстратов, прежде всего в клетках коры мозга, которое сохранялось через 96 часов, когда такие нарушения характеризовались более тяжелой формой в нейронах коры и гиппокампа. Актовегин (20 мг/мл) восстанавливал измененный баланс, наблюдаемый после ишемии/реперфузии в клетках коры мозга и гиппокампе и снижал их повреждающее влияние на клетки, что способствовало выживанию нейронов в этих экстремальных условиях [35].
Связь между HIF-1 и митоген- активируемой киназой р38
В исследовании G. Semenza и соавт. установлена прямая связь между апоптозом, вызываемым ультрафиолетовым облучением (УФ) кератиноцитов человека, и стимуляцией протеин киназы р38МАРК, которая через HIF-1-фактор транскрипции генов и проапоптозный белок Вах контролирует выход цитохрома С из митохондрий в цитоплазму, индуцирующий каскад каспаз, ответственных за разрушение ДНК и формирование апоптоза [36]. Таким образом, сигнальный путь р38МАРК/HIF-1 играет центральную роль в регуляции гибели клеток кожи человека, индуцированной УФ. В одной из наших последних работ на клетках нейробластомы человека линии SK-N-SH in vitro было изучено влияние Актовегина на внутриклеточные сигнальные пути, связанные с участием митоген-активируемой киназы (р38МАРК); киназами, регулируемыми влиянием внеклеточных стимулов на рецепторы (ERKs); фосфатидилинозитол-3 киназой (PI-3K) и c-Jun-N-SH терминальной киназой (JNK) c использованием селективных ингибиторов. Показано, что Актовегин защищал клетки линии SK-N-SH от апоптоза, индуцированного пероксидом водорода (Н2О2); этот эффект был связан с влиянием препарата на сигнальные пути р38МАРК и PI-3K [31]. Эти результаты свидетельствуют о том, что независимо от природы стимула, индуцирующего апоптоз (УФ или Н2О2), одну из главных ролей в этом процессе играет протеин киназа р38, которая контролирует активность HIF-1 не только в нормальных клетках (кератиноциты кожи), но и в сигнальном пути опухолевых перевиваемых нейронов человека линии SK-N-SH.
Заключение
Подводя итог, можно заключить, что ишемия и последующая гипоксия выступают в качестве универсального механизма, в значительной степени ответственного за выживание опухолевых клеток и поддержание их жизнеспособности в неблагоприятных условиях. Эти процессы тесно связаны с развитием ангиогенеза в сóлидных опухолях, изменением энергетического обмена, ответственного за усиление гликолиза, а также закислением цитоплазмы, блокирующим запуск эндогенного механизма апоптоза, опосредуемого митохондриями. Во всех этих процессах участвует активация фактора HIF-1, которая развивается при гипоксии. Увеличение поступления кислорода в опухолевые клетки вызывает деградацию этого кислород-зависимого фактора транскрипции многочисленных генов. В экспериментальных работах было показано, что подавление активации HIF-1 и его взаимодействия с ДНК, достигаемое с помощью различных воздействий, тормозит пролиферацию опухолевых клеток, снижает их метастазирование и жизнеспособность.
В развитии опухолей и их озлокачествлении участвует множество процессов и факторов, одновременное воздействие на которые с помощью поликомпонентных препаратов, по-видимому, должно оказывать более эффективный антиканцерогенный эффект. Одним из новых направлений в этой области является использование в качестве мишени HIF-1, для разрушения которого необходимо увеличить поступление в клетки кислорода с помощью поликомпонентных синтетических или природных антигипоксантов, включая Актовегин. Этот препарат усиливает поступление кислорода в клетки и митохондрии, активирует окислительное фосфорилирование в митохондриях, подавляет внутриклеточную генерацию радикалов кислорода, которые активируют радикал-зависимые сигнальные каскады, контролирующие активацию HIF-1.
Для проверки этого предположения необходимо проведение дополнительных исследований, направленных на выявление уровня ключевых маркеров активности HIF-1 и жизнеспособности опухолевых клеток, подвергшихся длительному воздействию Актовегина в условиях in vitro и in vivo.


