
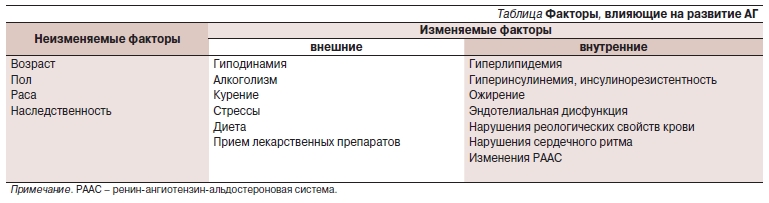
Артериальная гипертензия (АГ) занимает одно из ведущих мест в структуре заболеваемости и смертности. По данным ВОЗ, АГ страдает от 20 до 40 % населения развитых стран. Россия относится к странам с очень высокой распространенностью АГ: в середине 1990-х гг. этот показатель составлял 39,9 % среди мужчин и 41,1 % среди женщин [1]. Результаты мониторинга эпидемиологической ситуации по АГ, проводимого в рамках целевой федеральной программы “Профилактика, диагностика и лечение АГ в России”, показали, что за последние 10 лет ситуация практически не изменилась: в 2004 г. распространенность АГ по-прежнему составляла 36,9 % среди мужчин и 42 % среди женщин [2]. Кроме того, результаты работы демонстрируют рост распространенности АГ с возрастом: после 60 лет она достигает 60 %, а после 80 – приближается к 80 %. В 95 % случаев речь идет об эссенциальной АГ, в то время как симптоматическая АГ выявляется только у 5 % больных. Факторы, влияющие на развитие АГ, представлены в таблице.
Таблица. Факторы, влияющие на развитие АГ.
Рассматривая внешние и внутренние факторы, ответственные за развитие АГ, можно сформулировать направления для профилактического и лечебного воздействия как на АГ, так и на ее осложнения. Воздействие на внешние факторы возможно только при мотивации больного на борьбу с АГ. К сожалению, способность и желание больных, особенно мужчин, изменить образ жизни для устранения основных внешних факторов, влияющих на АГ, крайне низка [3]. Как в России, так и в мире число больных, контролирующих артериальное давление (АД), невелико: но если во Франции, Греции, Германии, Испании и США целевой уровень АД достигается у 30,0–35,7 % больных АГ, то в Канаде, Чехии, Польше, Индии и России эта величина не превышает 9–16 % [4].
Причин низкой приверженности больных к гипотензивной терапии множество:
• плохая переносимость гипотензивных препаратов;
• низкая дисциплинированность больных;
• высокая стоимость лекарств;
• психологические факторы (нежелание принимать препараты пожизненно);
• недостаточная осведомленность об осложнениях АГ и целях приема препаратов;
• уже имеющийся когнитивный дефицит – снижение памяти (пациенты забывают принимать препарат), рассеянность (нерегулярность приема лекарств), снижение критики к своему состоянию;
• антигипертензивные препараты не устраняют многие неврологические симптомы (головокружение, головная боль, неустойчивость при ходьбе, тревожность, раздражительность), характерные для поражения головного мозга при АГ.
Систематические нарушения в гипотензивной терапии или отказ от нее приводят к снижению эффективности лечения и усугублению АГ с развитием осложнений со стороны органов-мишеней [5].
Вследствие АГ происходит поражение сердца, сосудов, почек, головного мозга. Одно из наиболее опасных осложнений АГ – острое нарушение мозгового кровообращения. Как впервые возникший, так и повторный инсульт является непосредственной причиной смерти больного в 57 % случаев на ранних сроках и в 14 % – в отдаленном периоде [6]. Также очень остро в современной неврологии стоит вопрос хронического поражения головного мозга вследствие АГ, в частности гипертензивной энцефалопатии (ГЭ). Данный термин еще в конце 1950-х гг. сотрудниками Института неврологии Максудовым Г.А. и Шмидтом Е.В. и определен как неуклонно прогрессирующее поражение вещества головного мозга, которое связано с плохо контролируемым повышением АД, ведущим к хронической ишемии мозга.
Среди факторов риска развития ГЭ выделяют следующие: неконтролируемая АГ, гипертонические кризы, высокая вариабельность АД, высокая ночная гипертензия, чрезмерное снижение АД, включая ятрогенное [7].
В течении ГЭ можно выделить три стадии. При I стадии в клинике доминируют субъективные нарушения в виде общей слабости и утомляемости, эмоциональной лабильности, нарушений сна, снижения
памяти и внимания, головных болей. Неврологическая симптоматика формируется не отчетливыми неврологическими синдромами, а представлена анизорефлексией, дискоординацией, симптомами орального автоматизма. Нарушения памяти, праксиса и гнозиса удается, как правило, выявить только при проведении специальных тестов.
При II стадии становится больше субъективных жалоб, а неврологическая симптоматика уже может быть разделена на отчетливые синдромы (пирамидный, дискоординаторный, амиостатический, дисмнестический), причем обычно доминирует какой-то один неврологический синдром. Профессиональная и социальная адаптация больных снижается. При III стадии нарастает неврологическая симптоматика, появляются отчетливый псевдобульбарный синдром, иногда пароксизмальные состояния (в т. ч. эпилептические припадки); выраженные когнитивные нарушения приводят к нарушению социальной и бытовой адаптации, полной потере работоспособности. Таким образом, ГЭ в конечном итоге приводит к формированию сосудистой деменции, причем зачастую формируется субкортикальный ее вариант [18].
При II и III стадиях ГЭ диффузные изменения вещества головного мозга обычно сочетаются с очаговыми поражениями в виде лакунарных инфарктов – небольших полостей размером от 0,1 до 1,0 см, образующихся в местах очагов ишемии мозга. В структуре цереброваскулярных заболеваний наблюдается увеличение доли лакунарных инсультов: среди всех случаев инсульта при АГ они составляют 15 %. Возможно либо бессимптомное развитие лакунарного инфаркта, если очаги находятся в функционально немых зонах, либо формирование транзиторной ишемической атаки, инсульта. Лакунарные инфаркты локализуются в базальных ядрах, внутренней капсуле, таламусе,
мосте, мозжечке и белом веществе полушарий. Образование множественного мелкоочагового пораже ния головного мозга – лакунарного состояния – значительно ухудшает прогноз течения ГЭ и снижает возможности медикаментозного улучшения состояния больных – даже при достижении хорошего контроля АГ. Кроме того, лакунарные инсульты являются факторами риска развития геморрагического инсульта и сосудистой деменции [19].
При АГ происходят следующие патоморфологические изменения в сосудах головного мозга: плазматическое и геморрагическое пропитывание, некроз стенки сосудов с ее последующим истончением, адаптивное утолщение стенок экстрацеребральных сосудов. При ГЭ наблюдается раннее поражение преимущественно белого вещества головного мозга, представляющее собой деструкцию миелина центральных проводников, имеющую типичную картину при компьютерной томографии (снижение интенсивности сигнала) и магнитно-резонансной томографии (повышение интенсивности сигнала) [8]. Характерные изменения выявляются обычно в зонах терминального кровоснабжения, особо чувствительных к колебаниям АД, т. е. в околовентрикулярных участках головного мозга, и называются лейкоареозом [9, 10]. При неконтролируемой АГ происходит прогрессирование описанных процессов в белом веществе, развивается феномен корковоодкоркового разобщения (в белом веществе локализуются проводящие волокна), нарушаются интеллектуально-мнестические функции и в итоге формируется сосудистая деменция (“субкортикальная артериолосклеротическая энцефалопатия”, или болезнь Бинсвангера). В исследовании ARIC показано, что подобные изменения белого вещества головного мозга встречаются в группе пациентов с АГ в 2 раза чаще, чем у нормотоников, а у больных с неконтролируемой АГ по сравнению с пациентами с адекватным контролем АГ – в 1,5 раза чаще. Нередко такие изменения протекают без клинической симптоматики. Продолжительность бессимптомной фазы может быть различной и определяется наличием других факторов риска [11].
При АГ интенсивно прогрессирует атеросклероз. Нарушаются структурно-функциональные свойства эритроцитов и тромбоцитов – ухудшается их способность к деформации, повышается гематокрит, увеличивается вязкость крови, что в свою очередь приводит к нарушению микроциркуляции. Патоморфологические, а также единичные клинические исследования венозной системы головного мозга при АГ свидетельствуют о выраженных нарушениях вплоть до облитерации венозных синусов мозга. В этом случае говорят об энцефалопатии смешанного генеза: гипертонической и атеросклеротической [12, 13].
Важным следствием изменений структуры сосудистой стенки является нарушение ауторегуляции мозгового кровообращения – способности поддерживать стабильный кровоток при колебаниях среднего системного АД в пределах от 60 до 150–170 мм рт. ст. [14]. Экспериментальные данные свидетельствуют о том, что расстройства ауторегуляции в первую очередь наблюдаются в белом веществе головного мозга и в меньшей степени – в коре больших полушарий [15]. В результате развивается гипоксия, обусловленная несоответствием между потребностью тканей в кислороде и его доставкой.
На начальном этапе кислородного голодания в митохондриях снижается скорость аэробного окисления и окислительного фосфорилирования, что приводит к уменьшению синтеза белков и экспрессии генов, снижению количества аденозинтрифосфата (АТФ), увеличению содержания аденозиндифосфата (АДФ) и аденозинмонофосфата. При дальнейшем снижении мозгового кровотока активируется фермент фосфофруктокиназа, усиливается анаэробный гликолиз, а затем происходит окончательный переход на анаэробное дыхание, что адаптирует клетку к гипоксии, однако запасы гликогена истощаются. Это влечет в свою очередь накопление недоокисленного лактата с развитием лактатацидоза. При этом увеличивается активность лактатдегидрогеназы и уменьшается активность сукцинатдегидрогеназы, поставляющей электроны в дыхательную цепь митохондрий, что указывает на нарушение процессов энергообразования в ишемизированном мозге. В таких условиях анаэробного гликолиза не происходит, что приводит к тяжелому энергодефициту. В конечном итоге возникают дестабилизация клеточных мембран, нарушение работы ионных каналов, повреждение калий-натриевого насоса, что приводит к гипергидратации клеток, мутному набуханию, а затем – к баллонной дистрофии. Важнейшая роль в этом процессе принадлежит глутаматным рецепторам.
Оксидантный стресс, тесно связанный с ишемическим каскадом, возникает при возбуждении глутаматных рецепторов и заключается в избыточном накоплении свободных радикалов, активации перекисного окисления липидов и избыточном внутриклеточном накоплении их продуктов. Реакции оксидантного стресса и ишемического каскада взаимодействуют и потенцируют друг друга.
Свободными радикалами являются высокоактивные формы кислорода, пероксид водорода, альдегиды, образующиеся в условиях гипоксии. Под их воздействием изменяются функциональные свойства ряда ферментов, углеводов, белков, в т. ч. ДНК и РНК; в результате клетка утрачивает свои функции, появляются аномальные белки и стимулируются вторичные деструктивные процессы [16–18].
Наряду с процессами свободнорадикального окисления в биологических объектах вырабатываются стабильные антиоксидантные радикалы или антиоксиданты. Механизм их действия основан на торможении свободнорадикальных процессов в тканях, что тормозит развитие деструктивных изменений, инактивирует реакции оксидантного стресса. От соотношения активности свободных радикалов и антиоксидантов зависят изменения структуры и функции субстратов, находящихся в условиях ишемии и стресса.
В организме имеется физиологическая антиоксидантная система, сохраняющая окислительно-антиоксидантное равновесие как в жидких средах (кровь, лимфа, внутриклеточная и межклеточная жидкость), так и в структурных элементах клетки (плазматических, эндоплазматических, митохондриальных, клеточных мембранах). К ферментным антиоксидантам относятся: супероксиддисмутаза, каталаза, глутатиондегидроаскорбатредуктаза, некоторые другие пероксидазы.
К неферментным антиоксидантам относятся витамины С, Е, К, глюкоза, убихиноны, фенилаланин, трансферрин, гаптоглобин, триптофан, церулоплазмин, каротиноиды. Биологические и химически синтезированные антиоксиданты делятся на жиро- и водорастворимые. Первые локализуются там, где расположены субстраты-мишени для атаки свободных радикалов и пероксидов, наиболее уязвимые для процессов перекисного окисления биологические структуры, к которым относятся прежде всего биологические мембраны, липопротеины крови (основными мишенями в них являются ненасыщенные жирные кислоты). Наиболее значимый жирорастворимый антиоксидант – α-токоферол. Среди водорастворимых антиоксидантов наиболее важны глутатион и система аскорбиновой кислоты, особенно значимая для антиоксидантной защиты мозга [21, 22].
На всех стадиях ГЭ к лечению основного заболевания – АГ – необходимо добавлять препараты, улучшающие мозговой кровоток и метаболизм нервной ткани, а также действующие на внутренние факторы, способные повлиять на прогрессирование энцефалопатии. С целью предупреждения нарастания атеросклеротического процесса необходима нормализация жирового обмена: снижение индекса массы тела, диета с низким содержанием жира, назначение статинов (симвастатина, правастатина). С целью улучшения реологических свойств крови показаны антиагреганты (ацетилсалициловая кислота, тиклопедин, клопидогрел, дипиридамол). В исследованиях CAPRIE и
ESPS-2 показано, что применение ацетилсалициловой кислоты, клопидогрела и дипиридамола уменьшает риск развития мозговых ишемий [20]. На всех стадиях ГЭ применение препаратов, действующих на сосудистую стенку, улучшающих метаболизм мозговой ткани и обладающих нейропротективными свойствами, является обязательным.
Исходя из патогенетической роли гипоксии и оксидантного стресса, в качестве одного из наиболее перспективных методов терапии хронических форм нарушений мозгового кровообращения, в т. ч. ГЭ, рассматривается применение антиоксидантов, являющихся специфическими корректорами энергетического метаболизма мозга.
В настоящее время в клинической практике применяются α-токоферол, аскорбиновая кислота, метионин, церуллоплазмин, каротин, убихинон, эмоксипин. Однако недостатком этих препаратов является необходимость длительного использования (в течение нескольких недель) для достижения в конечном итоге слабовыраженного антиоксидантного и антигипоксантного эффектов. Это обусловило предпосылки для создания более эффективных лекарственных средств с антиоксидантными свойствами, среди которых заметное место занимает Мексидол® [19].
Мексидол – производное 3-оксипиридина, относящееся к водорастворимым антиоксидантам биогенного типа и являющееся структурным аналогом соединений группы витамина В6. Важным свойством этого препарата является способность проникать через гематоэнцефалический барьер. Препарат Мексидол (2-этил6-метил-3-гидроксипиридина сукцинат) был создан на основе эмоксипина с включением в его молекулу янтарной кислоты.
В последние годы широко изучается действие янтарной кислоты, ее солей и эфиров, представляющих собой универсальные внутриклеточные метаболиты. Янтарная кислота содержится во всех тканях и органах. Выполняя каталитическую функцию по отношению к циклу Кребса, это соединение снижает в крови концентрацию других продуктов цикла – лактата, пирувата, цитрата, продуцируемых и накапливающихся на ранних стадиях гипоксии, и тем самым включается в энергетический обмен,
направляя процесс окисления по наиболее экономичному пути. В нервной ткани функционирует цикл Робертса, в ходе которого из γ-аминомасляной кислоты (ГАМК) через промежуточную стадию янтарного альдегида образуется янтарная кислота. Образование янтарной кислоты возможно также в условиях гипоксии и окислительного стресса в реакции окислительного дезаминирования α-кетаглутаровой кислоты в печени. Антиоксидантное действие янтарной кислоты связано с ее влиянием на транспорт медиаторных аминокислот, а также с увеличением содержания в мозге аминомасляной кислоты за счет шунта Робертса. Янтарная кислота в организме нормализует содержание медиаторов воспаления гистамина и серотонина, повышает микроциркуляцию в органах и тканях, прежде всего в мозге, не оказывая влияния на АД и показатели работы сердца. Антигипоксантный эффект янтарной кислоты связан с активацией сукцинатдегидрогеназного окисления и восстановлением активности цитохромоксидазы – ключевого окислительно-восстановительного фермента дыхательной цепи.
Соответственно, исходя из химической формулы, Мексидол является антиоксидантом, мембранопротектором, антигипоксантом прямого энергизирующего действия, ингибирующим свободные радикалы, уменьшающим активацию перекисного окисления липидов, повышающим активность собственной физиологической антиоксидантной системы, активирующим энергосинтезирующие функции митохондрий и улучшающим энергетический обмен в клетке. Мексидол оказывает модулирующее влияние на мембрансвязанные ферменты, ионные каналы, рецепторные комплексы, в т. ч. ГАМК и ацетилхолиновые, улучшает синаптическую передачу в мозговых структурах, корригируя расстройства в микроциркуляторных системах. Мексидол действует в условиях ишемии и гипоксии как специфическая ловушка свободных радикалов, снижая их повреждающее действие на церебральные структуры
[23]. Препарат назначают в дозах от 200 до 500 мг 2–4 раза в сутки внутривенно струйно или капельно на физиологическом растворе или внутримышечно. Максимальная суточная доза – 1200 мг. Затем возможен переход на таблетированную форму приема. Таблетка Мексидола содержит 125 мг этилметилгидроксипиридина сукцината; назначается препарат внутрь по 125–250 мг 3 раза в сутки. Начинают лечение с дозы 125–250 мг 1–2 раза в сутки с постепенным повышением до получения терапевтического эффекта. Длительность лечения обычно составляет 2–6 недели. Лечение прекращают постепенно, уменьшая дозу в течение 2–3 дней.
У больных с ГЭ длительная плановая терапия антиоксидантами значимо улучшает качество жизни и позволяет предотвращать прогрессирование функциональноморфологических церебральных расстройств. Ранняя терапия антиоксидантами в настоящее время рассматривается как патогенетически обусловленный метод коррекции церебрального метаболизма при гипертензивной энцефалопатии.
ГЭ – тяжелое прогрессирующее заболевание, формирующее разнообразные неврологические синдромы, угрожающее развитием инсультов и приводящее к сосудистой деменции. Своевременно начатое лечение может на долгие годы сохранить профессиональную, социальную и бытовую адаптацию больного, улучшает прогноз в отношении продолжительности жизни больного.



{kind=link}