
Цирроз печени (ЦП) является финальной стадией диффузных воспалительных процессов печени и характеризуется нарушением ее долькового строения в результате прогрессирующего фиброза и образования узлов регенерации. В экономически развитых странах ЦП входит в число шести основных причин смерти пациентов от 35 до 60 лет, составляя 14–30 случаев на 100 тыс. населения. В связи с ростом заболеваемости хроническими вирусными гепатитами и метаболическими поражениями печени в ближайшее десятилетие ожидается существенное увеличение числа больных ЦП.
Причины развития ЦП многообразны [1] и основными этиологическими факторами являются следующие:
• Вирусы гепатита В, С, D (HBV, HCV, HDV соответственно).
• Алкоголь (прием этанола > 80 г в сутки, ≥ 5 раз в неделю в течение > 5 лет).
• Тезаурисмозы – болезни накопления (гемохроматоз, болезнь Вильсона–Коновалова, недостаточность α1-антитрипсина и др.).
• Неалкогольный стеатогепатит и гепатит (лекарственные, метаболические и др.).
• Аутоиммунный гепатит.
• Холестаз внутри- и внепеченочный (длительно существующий).
• Нарушение венозного оттока от печени (синдром Бадда–Киари, венооклюзионная болезнь, констриктивный перикардит и др.).
• Неуточненная этиология (криптогенный цирроз).
Диагностика ЦП основывается на результатах клинико-инструментальных и морфологических исследований [1, 2]. Морфологическая картина при ЦП характеризуется наличием фиброзных септ и ложных долек. В зависимости от размеров ложных долек выделяют мелкоузловой (узлы регенерации до 3 мм в диаметре), крупноузловой (3 мм и более) и смешанный ЦП [3]. Для оценки тяжести воспалительно-некротических изменений паренхимы печени и стадии фиброза предложено несколько систем. В последнее время предпочтение отдается системам Knodell в модификации K. Ishak и соавт. и METAVIR, позволяющих проводить полуколичественную оценку степеней активности воспаления и стадии фиброза при хронических заболеваниях печени. Тем не менее морфологический метод имеет ряд недостатков: инвазивность, возможность развития осложнений при пункционной биопсии, высокую вариабельность в трактовке морфологической картины различными исследователями, отсутствие единой унифицированной системы трактовки результатов гистологического исследования, наличие у большинства пациентов с ЦП противопоказаний к проведению пункционной биопсии печени и др. [4, 5].
В настоящее время в оценке степени активности и стадии фиброза в качестве альтернативы морфологическому исследованию используются неинвазивные методы [6]. К ним относятся биопрогностические лабораторные тесты (ФиброТест, АктиТест, ФиброМакс системы и др.), эластометрия и магниторезонансная эластография, ультразвуковое исследование печени, селезенки и сосудов портальной системы кровотока, компьютерная, магниторезонансная томографии печени и др. Достоинством данных методов является возможность проводить динамическое наблюдение за течением заболевания и контролировать эффективность лечения.
Клинические проявления ЦП обусловлены печеночно-клеточной недостаточностью, синдромом портальной гипертензии и ее осложнениями, а также системными поражениями, связанными с действием этиологического фактора, иммунологических и метаболических расстройств [7–9]. Частота выявления и степень выраженности клинических симптомов зависят от стадии развития, компенсации, а также частично от этиологии ЦП, увеличиваются с нарастанием продолжительности заболевания.
Основными проявлениями печеночно-клеточной недостаточности являются астеновегетативный синдром (слабость, утомляемость и др.); желтуха; вазодилатация и гипердинамический тип кровообращения (низкое артериальное давление, тахикардия, снижение церебрального почечного и печеночного кровотока); печеночная энцефалопатия; кожные (сосудистые звездочки, печеночные ладони и др.) и эндокринные изменения; нарушение свертывания крови. О наличии портальной гипертензии свидетельствуют спленомегалия, расширение вен пищевода, кардиального отдела желудка, передней брюшиной стенки и аноректальных вен; выявление при ультразвуковом и рентгенологическом исследованиях увеличения диаметра воротной и селезеночной вен, а также портакавальных коллатералей. Осложнения портальной гипертензии включают асцит, или отечно-асцитический синдром, кровотечения из варикознорасширенных вен пищевода, желудка и аноректальных вен, печеночную энцефалопатию, гепаторенальный синдром, гиперспленизм и гипертензионную портальную гастро-, энтеро- и колонопатию.
По механизму развития различают следующие системные проявления при хронических заболеваниях печени, включая ЦП [10]:
• антигенстимулированные иммунные процессы, включающие криоглобулинемию II типа (синовиит, васкулит, периферическую невропатию, синдром Рейно), гломерулонефрит, кожные васкулиты;
• аутоантигензависимые иммунные процессы – аутоиммунный тиреоидит, синдром Съегрена, болезнь Грейвса, красный плоский лишай, лихорадка, полиартралгии, артриты, лимфаденопатия, васкулиты, цитопении;
• процессы, обусловленные действием этиологического фактора на другие органы и системы.
Нередко, особенно на ранних стадиях развития ЦП, в клинической картине системные проявления могут быть превалирующими.
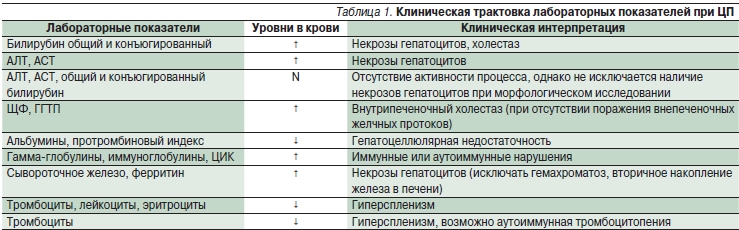
Лабораторные исследования при ЦП включают клинический анализ крови и комплекс биохимических тестов, позволяющих оценивать состояние синтетической функции гепатоцитов, степень активности процесса, выявлять холестаз, иммунологические и гематологические нарушения, требующие соответствующей терапии (табл. 1).
Примечание. Уровень в крови: ↑ – повышен, ↓ – снижен, N – нормальный. АЛТ – аланинаминотрансфераза, АСТ – аспарагинаминотрансфераза, ГГТП – гамма-глутамилтранспептидаза, ЩФ – щелочная фосфатаза, ЦИК – циркулирующие иммунные комплексы.
При анализе биохимических проб печени с определенной долей вероятности возможно установить ведущие механизмы цитолиза гепатоцитов, что определяет тактику патогенетической терапии. Патогенетические варианты цитолитического синдрома при хронических заболеваниях печени включают:
• Некрозы гепатоцитов, обусловленные накоплением в клетках свободных радикалов и перекисей, активизацией перекисного окисления липидов (ПОЛ) клеточных мембран без холестаза и аутоиммунных нарушений. Отличительные признаки: повышение уровней АЛТ, АСТ, нормальный уровень ЩФ, гамма-глобулинов, возможно повышение уровня ГГТП.
• Некрозы гепатоцитов с интралобулярным (гепатоцеллюлярным и каналикулярным) холестазом. Отличительные признаки: повышение уровней АЛТ, АСТ, ГГТП в сочетании с повышением уровня ЩФ до 2 норм.
• Некрозы гепатоцитов с экстралобулярным (дуктулярным) холестазом. Отличительные признаки: повышение уровней АЛТ, АСТ, ГГТП в сочетании с повышением уровня ЩФ более 2 норм.
• Некрозы гепатоцитов аутоиммунного генеза. Отличительные признаки: повышение уровней АЛТ, АСТ, гамма-глобулинов и/или иммуноглобулинов G в 1,5 и более раз.
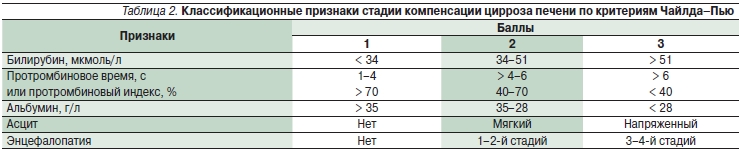
Для оценки степени компенсации заболевания используются критерии Чайлда–Пью (табл. 2).
Примечание. Стадия компенсации ЦП по Чайлд–Пью: ЦП класс А (компенсированный) – 5–6 баллов, ЦП класс В (субкомпенсированный) – 7–9 баллов, ЦП класс С (декомпенсированный) – 10–15 баллов.
Гепатоцеллюлярные некрозы, воспаление и фиброз являются взаимосвязанными процессами, персистенция которых лежит в основе прогрессирования ЦП. Печеночные клетки могут быть повреждены непосредственным воздействием этиологических агентов, таких как алкоголь, вирус, или являются объектом аутоиммунной и иммунной агрессии, а также компонентов клеточных некрозов и воспаления (провоспалительных цитокинов и др.). Цитолиз гепатоцитов нередко обусловлен накоплением в них желчных кислот и других токсических компонентов желчи при наличии внутрипеченочного холестаза, а также развитием ишемии в центральных зонах ложных долек в результате снижения кровотока, уменьшения количества венозных и артериальных сосудов и капилляризации оставшихся синусоидов на периферии ложных долек. Определенную роль в формировании некрозов гепатоцитов играют иммунологические нарушения, обусловленные дисфункцией купферовских клеток, синтез провоспалительных цитокинов (фактора некроза опухоли α, интерлейкинов 1, 6 и др.), а также эндотоксемия, которая при ЦП связана в первую очередь с развитием избыточного бактериального роста в кишечнике, транслокацией кишечных бактерий и их токсинов в систему воротной вены, лимфу и поступлением их в печень.
Вторым чрезвычайно важным механизмом формирования и прогрессирования ЦП является избыточная продукция фибриллярной соединительной ткани. Продуцентами экстрацеллюлярного матрикса печени (ЭМП) – основного компонента внутрипеченочной соединительной ткани – являются три типа клеток, входящих в структуру стенок синусоидов: стеллатные клетки (экспрессируют все составляющие матрикса, включая коллагены, гликопротеины и протеогликаны), гепатоциты (синтезируют ламинин и коллаген XVIII типа) и эндотелиоциты (секретируют фибронектин и коллаген IV типа). Ведущая роль как в синтезе, так и в деградации компонентов ЭМП принадлежит стеллатным клеткам, которые в нормальной печени находятся в покоящемся состоянии, постоянно продуцируя определенное количество ЭМП, а также несколько типов металлопротеиназ, участвующих в его деградации (протеолизе), что обеспечивает нормальный количественный и качественный состав внутрипеченочной соединительной ткани.
При ЦП в зонах повреждения постоянно присутствует множество биологических субстанций, которые непрерывно активируют стеллатные клетки, что приводит к трансформации их в миофибробласты, непрерывно продуцирующие фибриллярный компонент соединительной ткани с формированием соединительнотканных септ и тяжей. К активаторам коллагенпродуцирующих клеток, которые являются потенциальными объектами лекарственной терапии, относятся трансформирующий фактор роста β1, тромбоцитарный фактор роста, факторы роста соединительной ткани и фибробластов, а также субстраты ПОЛ и другие субстанции некротизированных гепатоцитов, цитокины, продуцируемые клетками Купфера, гепатоцитами, лейкоцитами, тромбоцитами; ацетальдегид, избыточное содержание железа в ткани печени и др. [11]. Накопления коллагена в пространствах Диссе, капилляризация синусоидов, последующее формирование соединительнотканных септ и тяжей приводят к развитию клинически значимых нарушений функций гепатоцитов и портальной гипертензии.
Таким образом, основные звенья патогенеза ЦП как точки приложения медикаментозной терапии включают:
• Персистирующее действие этиологического фактора.
• Повышение функциональной нагрузки на гепатоциты – накопление в печеночных клетках эндогенных и экзогенных токсических субстратов, ведущих к усилению ПОЛ, нарушению стабильности клеточных мембран.
• Формирование внутрипеченочного холестаза – задержка в гепатоцитах компонентов желчи, приводящая к их некрозу.
• Нарушение кровоснабжения паренхимы печени (узлов регенерации) – развитие ишемических некрозов гепатоцитов за счет капилляризации синусоидов и уменьшения сосудистого русла; повышение потребления кислорода для обеспечения метаболизма гепатоцитов.
• Повышение мезенхимальной активности, обусловленное нарушением клиренса бактериальных антигенов и формированием аутоантигенов, что запускает иммунный и аутоиммунный механизмы некрозов гепатоцитов.
• Прогрессирование фиброза, связанное с постоянной мультифакторной активацией стеллатных клеток.
При ЦП развивается ряд метаболических нарушений, лежащих в основе самопрогрессирования заболевания даже при удалении этиологического фактора, отягощающих его течение и требующих терапевтических вмешательств [1]. Так, у пациентов с ЦП рано формируется отрицательный энергетический баланс. Известно, что источником энергии, необходимой для обеспечения гепатоцеллюлярного метаболизма, является оксидация короткоцепочечных жирных и аминокислот в митохондриях. Данный процесс приводит к резкому падению парциального давления кислорода в центролобулярной и перипортальной зонах, что при ЦП является фактором риска развития гипоксических некрозов гепатоцитов в связи с нарушением печеночного кровотока.
При этом следует учитывать, что для больных ЦП основным источником короткоцепочечных жирных и аминокислот является катаболизм жировой и мышечной тканей, что сопровождается снижением массы тела, атрофией мышц и в конечном итоге – развитием кахексии.
При ЦП наблюдается три типа расстройств углеводного обмена:
1. Гипогликемия.
2. Гепатогенный сахарный диабет.
3. Нарушение гликогенолизиса и глюконеогенеза.
Гипогликемия развивается довольно редко. В ее основе лежит редукция глюконеогенеза в гепатоцитах, связанная с уменьшением объема функционирующей паренхимы. При ведении больных ЦП следует учитывать, что употребление алкоголя может провоцировать развитие гипогликемии. Сахарный диабет и снижение толерантности к глюкозе выявляются среди 10–15 % и 50–80 % пациентов соответственно. В основе их развития лежит периферическая инсулинорезистентность, которая развивается на фоне гиперинсулинемии, обусловленной нарушением деградации инсулина в печени и повышением его секреции, стимулированной гипергликемией. При ЦП отмечено существенное снижение содержания гликогена в печени и как следствие – гликогенолиза, связанное с редукцией количества гепатоцитов, печеночного кровотока, а также с наличием портакавальных анастомозов, ограничивающих поступление глюкозы в портальную вену.
В результате нормальный уровень глюкозы поддерживается компенсаторным увеличением глюконеогенеза, для обеспечения которого необходимы аминокислоты, основным источником которых является катаболизм протеинов мышечной ткани, что приводит к ее атрофии.
Нарушения белкового обмена включают три типа расстройств:
1. Снижение синтеза большинства протеинов, в первую очередь альбумина.
2. Дисбаланс аминокислот в плазме крови.
3. Снижение синтеза мочевины.
Превалирование катаболизма над синтезом протеинов при ЦП связано не только с уменьшением числа печеночных клеток, но и с повышением уровня катехоламинов в крови в результате симпатикотонии. Вследствие повышенного катаболизма протеинов мышечной ткани, снижения расщепления в печени ароматических аминокислот, дефицита S-аденозилметионинсинтетазы развивается дисбаланс аминокислот в плазме крови, нарушается синтез глутатиона, полиаминов, фосфолипидов, нейротрасмиттеров и медиаторов. В результате нарушаются детоксикационная функция печени, стабильность клеточных мембран, регенерация гепатоцитов. Снижение синтеза мочевины приводит к повышению содержания аммиака в плазме крови и метаболическому алкалозу, что является фактором риска развития печеночной энцефалопатии.
При ЦП выявляются зависящие от этиологии заболевания различные варианты нарушений липидного обмена, наиболее значимыми из которых являются гипертриглицеридемия, появление атипичных липопротеинов, снижение синтеза фосфолипидов. Уже на ранней стадии заболевания отмечается нарушение экскреции желчных кислот из гепатоцитов (внутриклеточный холестаз), которые оказывают цитотоксический эффект. В случае выраженного холестаза, приводящего к билиарной недостаточности, нарушается всасывание жирорастворимых витаминов с последующим дефицитом витаминов A, Д, E и К. Метаболические нарушения при ЦП усугубляются наличием дефицита как водо-, так и жирорастворимых витаминов, который связан с уменьшением их потребления с пищей, нарушением всасывания, приемом алкоголя, развитием кишечного дисбиоза и др. Заместительная терапия необходима даже в отсутствие клинических признаков гиповитаминоза.
Важная роль в патогенезе клинических проявлений, осложнений и прогрессировании ЦП принадлежит нарушению нормального cостава кишечной микрофлоры, которое выявляются среди 50–70 % пациентов [12–14].
В патогенеза развития кишечного дисбиоза при ЦП выделяются три ключевых звена:
1. Нарушение моторной функции кишечника.
2. Дефицит желчных кислот.
3. Развитие портальной гипертензионной энтеро- и колопатии.
Замедление тонкокишечного транзита с эпизодами интестинального стаза обусловлено дисбалансом циркулирующих гастроинтестинальных гормонов и пептидов в результате портосистемного шунтирования; развитием миопатии и периферической невропатии с нарушением сократительной функции гладкой мускулатуры кишечника; наличием асцита, который несомненно сопровождается развитием интестинального стаза. Дефицит желчных кислот в кишечнике как проявление печеночно-клеточной недостаточности и/или холестаза приводит к снижению бактерицидности желчи, замедлению моторики кишечника и билиарной системы, нарушению гидролиза липидов.
Развитие портальной гипертензионной энтеро- и колонопатии сопровождается нарушением кровотока и изменением структуры сосудистых стенок в слизистой оболочке кишки, повреждением кишечного слизистого барьера и транслокацией кишечной микрофлоры за пределы кишки, увеличением функциональной нагрузки на печень и нарастанием печеночно-клеточной недостаточности, развитием системной эндотоксемии. Наличие кишечного дисбиоза у больных ЦП является дополнительным фактором риска развития неалкогольной жировой дистрофии гепатоцитов, внутрипеченочного интралобулярного холестаза, печеночно-клеточной дисфункции, воспалительных и дискинетических нарушений внепеченочного билиарного тракта, а также ряда осложнений (асцита – перитонита, печеночной энцефалопатии, эпизодов бактериемии, бактериальных инфекций и др.).
Таким образом, при ведении больных ЦП большое значение имеет уточнение этиологии и ведущих патогенетических механизмов прогрессирования процесса и формирования симптомов заболевания, а также характера метаболических нарушений. Кроме того, важно установить степень компенсации заболевания и определить тот рубеж, когда консервативная терапия является малоэффективной и больной нуждается в пересадке печени.



{kind=link}
{kind=link}