
Введение
Гипертрофическая кардиомиопатия (ГКМП) является генетическим заболеванием миокарда с распространенностью 1 случай на 200–500 человек [1–3]. При этом заболевании идентифицировано множество патогенных мутаций генов, кодирующих различные белки саркомера [4, 5]. Наряду с генетической гетерогенностью у пациентов с ГКМП отмечается разнообразие клинических проявлений, фенотипов гипертрофии миокарда, течения и исходов заболевания [5, 6].
В последние десятилетия в лечении ГКМП применяются различные фармакологические препараты и нефармакологические подходы, включая методы септальной редукции (МСР), имплантацию кардиовертеров-дефибрилляторов и трансплантацию сердца [3, 5]. Несмотря на успехи в лечении, ГКМП является прогрессирующим заболеванием, при котором развитие фибрилляции предсердий и сердечной недостаточности (СН) наблюдается у каждого 5-го больного в возрасте 50–70 лет [7].
Согласно российским рекомендациям по ГКМП [1], при лечении больных обструктивной формой заболевания с наличием симптомов, ухудшающих повседневную активность и качество жизни, рекомендуется назначение β-адреноблокаторов (БАБ) и негидропиридиновых блокаторов кальциевых каналов (БКК). По рекомендациям Европейского общества кардиологов по лечению кардиомиопатий 2023 г. и Американской ассоциации сердца/Американской коллегии кардиологов по диагностике и лечению больных ГКМП в случаях, рефрактерных к терапии с помощью БАБ и БКК, можно назначать дизопирамид [3, 8–10].
Пациентам с наличием выраженных симптомов, обусловленных обструкцией выносящего тракта (ВТ) левого желудочка (ЛЖ; градиент >50 мм рт.ст.), несмотря на применение максимально переносимых доз лекарственной терапии, показано лечение с помощью МСР, к которым относятся хирургическая миоэктомия и алкогольная септальная аблация [11, 12].
Последние годы ознаменовались открытием нового поколения болезнь-модифицирующих препаратов, воздействующих на миокардиальную гиперконтрактильность и нарушения энергетического баланса благодаря аллостерическому ингибированию миозина – основного белка, генерирующего силу сердечной мышцы [13, 14]. Настоящий обзор посвящен обсуждению новой группы фармакологических препаратов в лечении ГКМП – ингибиторов кардиального миозина (ИКМ).
Структура и сокращение саркомера
Саркомер представляет собой основную сократительную единицу поперечно-полосатой мускулатуры, состоящую из толстых и тонких филаментов. Основные сократительные белки саркомера представлены миозином толстого и актином тонкого филамента [15, 16]. Тонкие филаменты состоят из нитей α-актина и кальций-чувствительного тропонин-тропомиозинового аппарата, который включает тропонин T, тропонин I, тропонин C и α-тропомиозин (рис. 1) [17, 18]. Тонкие филаменты соединены с Z-линиями на обоих концах саркомера. Саркомер ограничен с обеих сторон Z-линиями, которые вместе с тонкими филаментами создают своего рода клетку вокруг толстой нити миозина, проходящей от центра саркомера наружу по направлению к Z-линии, не достигая ее [19].

Толстые филаменты саркомера состоят из белков, обеспечивающих как моторные, так и регуляторные функции. Тяжелая цепь β-миозина (β-MHC – β-myosin heavy chain) содержит структурные и функциональные домены. Спиральные хвостики миозина собираются вместе, образуя цилиндрическую основу из толстых нитей, от которой через равные промежутки времени по спирали отходят пары миозиновых головок.
Головка миозина содержит нуклеотид-связывающий «карман» с активностью гидролазы АТФ, сайты связывания с актином и регуляторные домены, представленные регуляторной и основной легкими цепями [15, 20].
Цикл сокращения саркомера инициируется катионом кальция (Ca2+), который связывается с тропонином С для уменьшения ингибирующего влияния на тропониновый комплекс [19]. После связывания Ca2+ с тропонином C актин взаимодействует с головкой миозина. Сложное взаимодействие тропонина С с тропонинами I и T приводит тропомиозин в состояние «открытия» актинового участка, к которому может прикрепляться головка миозина. Во время сокращения головки миозина захватывают актин и тянут актиновые нити к центру саркомера. Таким образом, тонкие и толстые нити скользят друг против друга, сокращая саркомер и длину клетки [19].
Генетические изменения и патогенез ГКМП
В настоящее время идентифицировано более 2100 мутаций в 12 основных генах, кодирующих сократительные белки миофиламентов сердечного саркомера и Z-полосы [4, 21, 22]. Наиболее распространенными являются мутации генов β-миозина тяжелой цепи (MHY7) и миозин-связывающего протеина-С (MYBPC3), вместе составляющие 60–70% патогенных вариантов [20–22].
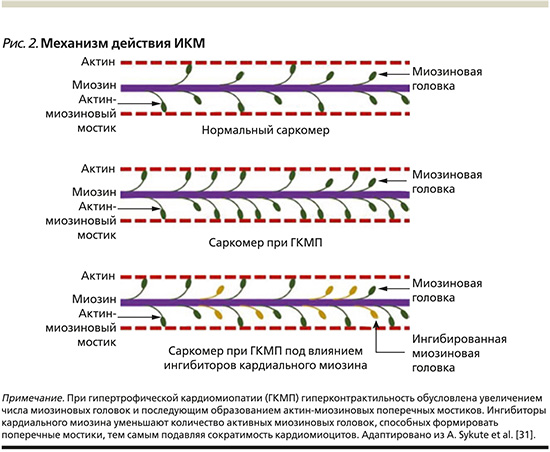
Любая мутация генов может изменять основные функции белков саркомера, включая сокращение, выработку энергии и утилизацию АТФ [23]. У пациентов с ГКМП отмечается неэффективность функции саркомера с повышенной чувствительностью миофиламентов к Са2+ и натрию [17]. Кроме того, мутации толстых филаментов приводят к увеличению числа миозиновых головок, которые, взаимодействуя с актином, приводят к состоянию гиперконтрактильности [24]. Мутации вызывают усиление метаболизма АТФ во время цикла образования актин-миозиновых мостиков, что приводит к истощению энергии и дисфункции миокарда [25, 26].
Ген, кодирующий MYBPC3, стабилизирует мотив взаимодействующих головок (англ.: interacting-heads motif) [27]. Вследствие мутаций в этом гене снижаются уровни белка, что дестабилизирует мотив взаимодействующих головок.
Экспериментальные исследования подтверждают эту гипотезу: в образцах ткани сердца мышей и человека с мутациями MYBPC3 обнаружено существенное снижение признаков суперрелаксации (SRX – super relaxation) и усиление состояния нарушенной релаксации (DRX – disordered relaxation) [26, 28, 29].
Мутации генов, обнаруживаемые при ГКМП, ответственны за нарушение свойств саркомеров и приводят к молекулярному фенотипу гипердинамической сократимости, нарушенной релаксации и повышенному потреблению энергии [29, 30]. Отмеченные фундаментальные нарушения проявляются уже на ранних стадиях у лиц с еще невыраженными фенотипическими признаками и приводят в дальнейшем к гипертрофическому ремоделированию [15, 22]. Многочисленные изменения на клеточном и молекулярном уровнях, наблюдаемые при ГКМП, представляют обоснованные мишени для болезнь-модифицирующей терапии.
Патофизиология
В результате дисрегуляции актин-миозинового взаимодействия нарушается контрактильная функция миокарда [31]. Патологически измененные белки саркомера приводят к развитию гипертрофии кардиомиоцитов, разнонаправленности мышечных волокон (англ. myocardial disarray), интерстициальному фиброзу и микрососудистой патологии [2, 18, 31]. Последующие патофизиологические события представлены обструкцией ВТЛЖ, диастолической дисфункцией, митральной регургитацией, миокардиальной ишемией и автономной дисфункцией сердца [32].
Традиционное фармакологическое лечение направлено на уменьшение выраженности клинических симптомов ГКМП благодаря применению БАБ и негидропиридиновых БКК [4, 33]. Поиск препаратов, нацеленных на образование актин-миозиновых мостиков, привел к созданию новой группы лекарственных веществ – ИКМ, представленных мавакамтеном и афикамтеном.
Мавакамтен
Мавакамтен (Mavacamten, MYK-461) является маломолекулярным, аллостерическим, обратимым ингибитором АТФ-азы кардиального миозина [34, 35]. Препарат действует на функцию гиперконтрактильности саркомеров, ингибирует избыточное образование актин-миозиновых поперечных мостиков, переводя общее количество миозина в энергосберегающее SRX (рис. 2) [10]. Мавакамтен замедляет скорость связывания миозина с актином как в АДФ (аденозиндифосфат)-связанном, так и в АДФ-свободном состоянии, в результате чего снижается сила сокращения саркомера и уменьшается сократимость миокарда [11].
Фармакокинетический профиль мавакамтена характеризуется отличной оральной биодоступностью (>85%) и быстрой абсорбцией со временем максимальной концентрации 1 час. Препарат имеет высокий объем распределения и длительную фазу выведения [11]. Прием пищи не оказывает значимого влияния на фармакокинетику препарата. Мавакамтен хорошо связывается с белками плазмы (97–98%). Отношение максимальной к минимальной концентрации мавакамтена в плазме крови в равновесном состоянии после приема один раз в сутки составляет ≈1,5 [36].
Средний период полувыведения – около 8 дней при нормальной функции ферментов системы цитохрома P450-CYP2C19. В отношении фармакодинамики отмечается дозозависимый характер снижения фракции выброса (ФВ) ЛЖ. Препарат имеет особое предостережение в плане риска развития СН и его не рекомендуется назначать пациентам с ФВЛЖ <55% [11].
Препарат выпускается в капсулах по 2,5 мг, 5,0, 10,0 и 15,0 мг для перорального применения. Перед началом терапии и в дальнейшем требуется мониторинг ФВЛЖ и градиента в ВТЛЖ с пробой Вальсальвы [31]. При значении ФВ<50% в любой период лечения необходимо прекратить прием мавакамтена. В случае прекращения терапии следует выполнять повторные эхокардиографические исследования каждые 4 недели до уровня ФВЛЖ>50%, после чего терапию можно возобновить со следующей более низкой дозой мавакамтена. Если ФВЛЖ пациента составляет <50% чаще 1 раза при дозировке 2,5 мг в сутки, терапия прекращается навсегда. Пациентам с нарушением функции печени или почек коррекция дозы не рекомендуется [31].
Афикамтен
Афикамтен (Aficamten, CK274, CK-3773274) – следующий в классе препарат группы ИКМ, который воздействует на миофиламенты саркомера и уменьшает число актин-миозиновых мостиков, приводя к дозозависимому снижению контрактильности миокарда [37–39]. Как и мавакамтен, афикамтен соединяется с АТФ-связывающим «карманом» миозина для стабилизации его в выключенное состояние и тем самым понижает сократительную способность миокарда.
Афикамтен был открыт в результате оптимизации соединения индолина. В отличие от мавакамтена он имеет более короткий период полувыведения, достигает состояние равновесия в течение 2 недель и может иметь более широкое терапевтическое «окно» [40]. Исследования показали, что афикамтен не обладает существенным воздействием на ингибирование или индукцию CYP450, обеспечивая меньше межлекарственных взаимодействий по сравнению с мавакамтеном [40].
Результаты клинических исследований ИКМ
К настоящему времени опубликованы результаты 4 клинических исследований, посвященных мавакамтену в лечении ГКМП (PIONEER-HCM, MAVERICK-HCM, EXPLORER-HCM и VALOR-HCM), и одного – афикамтену (REDWOOD-HCM) [34, 38, 41].
В исследовании PIONEER-HCM [42] первичной конечной точкой являлось уменьшение градиента в ВТЛЖ после физической нагрузки у больных ГКМП спустя 12 недель лечения мавакамтеном. Все пациенты были разделены на 2 группы: А – прекратившие прием всех лекарственных препаратов как минимум за 14 дней до начала исследования и В – продолжавшие принимать ранее назначенную терапию. В группе А начальная доза мавакамтена составляла 10–15 мг/сут в зависимости от массы тела пациента, в группе В – 2–5 мг/сут. Спустя 4 недели лечения доза могла быть увеличена в зависимости от изменений градиента ВТЛЖ и ФВЛЖ. Симптомы СН II функционального класса (ФК) по NYHA (New York Heart Association) имели 57% больных, III класса – 43%.
Исследование PIONEER-HCM достигло своей первичной конечной точки со снижением пикового градиента ВТЛЖ после физической нагрузки через 12 недель. Прием мавакамтена сопровождался статистически значимым уменьшением среднего градиента ВТЛЖ после физической нагрузки: в группе А со 103 до 19 мм рт.ст. (p=0,008), в группе В с 86 до 64 мм рт.ст. (p=0,020). Отмечено более выраженное снижение ФВЛЖ в группе А: в среднем на 15% (доверительный интервал [ДИ] от -23 до -6%) по сравнению с группой В – на 6% (ДИ от -10 до -1%). Препарат в целом хорошо переносился, в большинстве случаев отмечались незначительные симптомы, включая слабость, тошноту и одышку.
Эффективность и безопасность применения мавакамтена были изучены на больных необструктивной ГКМП в исследовании MAVERICK-HCM, в котором приняли участие 59 пациентов, разделенных на 2 группы, принимавшиех 2 разные дозировки мавакамтена и плацебо [43]. В исследование были включены пациенты с диагнозом ГКМП, значением градиента ВТЛЖ<30 мм рт.ст., симптомами СН II/III ФК по NYHA, повышенными уровнями натрийуретического пептида (NT-proBNP – N-terminal pro-B-type natriuretic peptide), значениями ФВЛЖ ≥55%.
Первичной целью была оценка безо-пасности и переносимости мавакамтена в течение 16-недельной терапии. Также была проанализирована комбинированная конечная точка, которая оценивала влияние исследуемого препарата на пиковое потребление кислорода (pVO2 – pulmonary venous oxygen tension) и ФК СН по NYHA. Серьезные побочные эффекты отмечены у 10% пациентов групп мавакамтена и у 21% − в плацебо группе. У 5 больных, получавших мавакамтен, наблюдалось обратимое снижение ФВЛЖ ≤45%. Среди пациентов из групп мавакамтена уровень NT-proBNP снизился на 53% по сравнению с 1% группы плацебо (p=0,0005), различия статистически значимы. Аналогичным образом выявлено снижение уровня тропонина I на 34% среди тех, кто принимал мавакамтен, по сравнению с увеличением на 4% в группе плацебо (p=0,009). Отмечена хорошая переносимость мавакамтена у большинства больных симптомной необструктивной ГКМП. Лечение мавакамтеном ассоциируется с существенным снижением уровней NT-proBNP и тропонина I, что свидетельствует об уменьшении напряжения стенки миокарда [43].
Исследование EXPLORER-HCM [34] было посвящено изучению эффективности и безопасности применения мавакамтена 251 пациентом с симптомами обструктивной ГКМП (градиент ВТЛЖ≥50 мм рт.ст.). Больные были рандомизированы в группы мавакамтена (n=123) или плацебо (n=128). Пациентам разрешалось принимать стандартную терапию по поводу ГКМП кроме дизопирамида и комбинированной терапии БАБ и БКК. Первичной конечной точкой являлось увеличение на ≥1,5 мл/кг/мин pVO2 или уменьшение выраженности ФК СН по NYHA на один и более, или увеличение pVO2 на ≥3,0 мл/кг/мин без ухудшения выраженности ФК по NYHA. Вторичные конечные точки включали изменения градиента ВТЛЖ после физической нагрузки, pVO2, ФК СН по NYHA, суммарного балла по Канзасскому опроснику для больных кардиомиопатией (Kansas City Cardiomyopathy Questionnaire-Clinical Summary Score) и балла по Симптомному опроснику одышки при ГКМП (Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness-of-Breath subscore). Первичная конечная точка была достигнута 37% пациентов группы мавакамтена по сравнению с группой плацебо – 17% (95% ДИ: 8,7–30,1; p=0,0005). По сравнению с группой плацебо у больных, получавших мавакамтен, отмечались более выраженные уменьшение градиента в ВТЛЖ (-47 против -10 мм рт.ст.; p<0,0001), увеличение значений pVO2 на ≥1,5 мл/кг/мин (37% против 17%; p=0,0006) и достоверное улучшение оценки симптомов по опросникам (p<0,0001). После лечения мавакамтеном у 65% больных отмечено уменьшение выраженности СН по сравнению с группой плацебо – 31% (p<0,0001).
В целом лечение мавакамтеном приводило к улучшению функциональной способности, уменьшению градиента обструкции и улучшению основных показателей самочувствия пациентов с ГКМП [34].
В исследовании VALOR-HCM [44] изучалась эффективность мавакамтена по оценке необходимости выполнения МСР у больных обструктивной ГКМП спустя 32 недели терапии. Исследование было разбито на несколько частей: 16-недельный плацебо-контролируемый период, 16-недельный период активной терапии мавакамтеном и продленный период, состоявший из 96 недель.
В исследование включали взрослых пациентов (средний возраст – 60,3 года), которых в течение последнего года направляли в центры для выполнения МСР при наличии выраженной одышки или боли в грудной клетке, несмотря на прием фармакологических препаратов в максимально переносимых дозировках, симптомов СН ФК III/IV по NYHA. В течение первых 16 недель пациенты были рандомизированы в группы плацебо или мавакамтена (5 мг/сут). Спустя 16 недель пациенты, получавшие плацебо, были переведены на терапию мавакамтеном. В течение всего периода доза мавакамтена титровалась каждые 4 недели на основе результатов эхокардиографической оценки ФВ и градиента ВТЛЖ.
Основным оцениваемым результатом являлось число пациентов, требующих выполнения МСР спустя 32 недели. К этому сроку 6 (10,7%) из 56 больных, изначально принимавших мавакамтен, и 7 (13,5%) из 52 больных перекрестной группы плацебо соответствовали рекомендуемым критериям для лечения МСР. Спустя 32 недели наблюдали устойчивое снижение градиента ВТЛЖ в покое (на 33 мм рт.ст.) и после пробы Вальсальвы (на 43 мм рт.ст.) в изначальной группе мавакамтена. Аналогичные показатели были отмечены в перекрестной группе плацебо–мавакамтен спустя 16 недель после приема мавакамтена. Отмечено также снижение выраженности СН в группе мавакамтена и перекрестной группе плацебо–мавакамтен. Таким образом, лечение мавакамтеном приводило к уменьшению пропорции больных, которым требовалось лечение с помощью МСР [44].
Опубликованы результаты одного рандомизированного плацебо‐контролируемого исследования REDWOOD‐HCM, посвященного эффективности и безопасности применения афикамтена. В исследовании принял участие 41 пациент с симптомной ГКМП с градиентом ВТЛЖ ≥30 мм рт.ст. в покое (или ≥50 мм рт.ст. после пробы Вальсальвы) [38]. Участники были разделены на 2 группы: получавшие афикамтен (n=28) и плацебо (n=13). Больные на базовой терапии были распределены на две подгруппы (по 14 человек) и получали афикамтен по 5 мг с последующим увеличением дозировки по 5 мг (до 15 мг/сут) или по 10 мг с увеличением по 10 мг (до 30 мг/сут).
Основной целью исследования являлась оценка безопасности и переносимости различных доз афикамтена больными симптомной обструктивной ГКМП по побочным эффектам и снижению ФВЛЖ<50%. Вторичные конечные точки включали изменение градиента ВТЛЖ, значения ФВЛЖ, уровней NT-proBNP, тропонина и выраженности СН.
Спустя 10 недель терапии афикамтеном у пациентов 1-й и 2-й подгрупп градиент в ВТЛЖ в покое снизился (средняя разница: -40±27 и -43±37 мм рт.ст., соответственно p=0,0003 и p=0,0004, по сравнению с плацебо) и после пробы Вальсальвы (-36±27 и -53±44 мм рт.ст.; p=0,001 и p=0,0001, по сравнению с плацебо). Наблюдалось умеренное снижение ФВ (-6,0±7,5% и -12,0±5,9%; p=0,007 и p=0,0001, по сравнению с плацебо). Отмечалось улучшение проявлений ФК СН по NYHA у 31% пациентов, получавших плацебо, и у 43 и 64% при приеме афикамтена в разных дозировках, но различия не были статистически значимыми. При применении афикамтена отмечалось снижение уровня NT-proBNP (на 62% по сравнению с плацебо, p=0,0002) [38].
Клиническое применение ИКМ
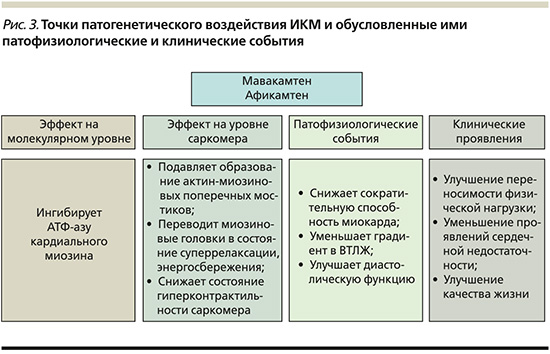
Инновационные методы лечения, действующие на специфические звенья патогенеза, расширяют терапевтические возможности в лечении ГКМП, позволяют улучшать выживаемость, качество жизни пациентов и уменьшать число осложнений [45]. ИКМ заслуживают обоснованного внимания в терапии больных ГКМП за счет воздействия на молекулярный субстрат заболевания с последующим благоприятным влиянием на патофизиологические события и клиническую картину заболевания (рис. 3).
Однако было бы неправильно рассматривать ИКМ в качестве конкурентов имеющимся методам лечения ГКМП. Они скорее дополняют арсенал возможных лечебных инструментов. Остается много открытых вопросов относительно ИКМ, включая уточнение их потенциальных преимуществ при лечении необструктивной формы ГКМП, индивидуальной вариабельности ответа среди пациентов различных групп и возможности в превентировании гипертрофической экспрессии у носителей патогенных мутаций саркомера [45]. Выполненные исследования проводились на пациентах, продолжавших принимать основную терапию, включая БАБ и БКК, поэтому отмеченные результаты ИКМ сложно воспринимать как обусловленные исключительно ими per se [46]. Более того, требует дальнейшего изучения отмеченный, пусть и невыраженный, отрицательный инотропный эффект ИКМ, что безусловно является существенным ограничением их применения, особенно в долгосрочном лечении.
В рекомендациях Совместного комитета Американской коллегии кардиологов и Американской ассоциации сердца 2020 г. препараты группы ИКМ не включены в качестве фармакологических средств при ГКМП [3].
В отсутствие результатов прямого сравнительного анализа Рабочая группа Европейского общества кардиологов в рекомендациях по лечению кардиомиопатий 2023 г. [10] не рекомендует использование ИКМ в качестве лекарственной терапии «первой линии», но рассматривает имеющиеся доказательства достаточно убедительными, чтобы обсуждать ИКМ как терапию «второй линии», если лечение с помощью БАБ, БКК и/или дизопирамида неэффективно или плохо переносится. Следует отметить, что дизопирамид не включен в российские клинические рекомендации по ГКМП 2020 г. [1], а сам препарат отсутствует в Государственном реестре лекарственных средств Российской Федерации.
ИКМ можно сочетать с БАБ или БКК. Повышение дозировки препарата до максимально переносимой следует проводить в соответствии с лицензированными рекомендациями под эхокардиографическим контролем.
В тех случаях, когда у пациентов имеются противопоказания или непереносимость БАБ, БКК или дизопирамида, ИКМ могут рассматриваться в качестве монотерапии [10].
Заключение
ИКМ представляют новый болезнь-модифицирующий класс препаратов для лечения больных ГКМП. Результаты клинических исследований показали, что эти препараты могут оказывать существенное влияние на уменьшение выраженности обструкции, замедление прогрессирования СН и улучшение качества жизни больных. Несмотря на отмеченные достоинства препаратов, необходимо продолжение дальнейших исследований по оценке безопасности ИКМ, изучению их эффективности вне основной терапии ГКМП и среди различных групп больных. Мавакамтен и афикамтен дополняют имеющийся арсенал лечебных подходов при ведении пациентов с ГКМП.


