
Термин “аутоиммунный панкреатит” (АП) был впервые предложен K.Yoshida и соавт. в 1995 г. применительно к хроническому панкреатиту (ХП), при котором были выявлены маркеры аутоиммунного воспаления [1]. Однако первое описание принадлежит H. Sarles и соавт. [2], которые наблюдали особый случай панкреатита с гипергаммаглобулинемией, затем были сообщения о сочетании ХП с другими аутоиммунными заболеваниями, такими как синдром Съегрена, первичный склерозирующий холангит (ПСХ) или первичный билиарный цирроз [3, 4]. Эти находки подтвердили гипотезу о том, что в основе ХП может лежать аутоиммунный механизм. Все эти данные позволили позднее выдвинуть концепцию аутоиммунного панкреатита как самостоятельного заболевания [1].
Основными признаками АП являются:
• повышенный уровень γ-глобулина или IgG в крови;
• наличие аутоантител;
• диффузное увеличение поджелудочной железы (ПЖ);
• диффузное неравномерное сужение главного панкреатического (вирсунгова) протока, иногда стеноз внутрипанкреатической части общего желчного протока, по данным ретроградной холангиопанкреатографии (РХПГ);
• фиброзные и воспалительные изменения в ткани ПЖ с преимущественной лимфоцитарной инфильтрацией паренхимы по данным морфологического исследования;
• бессимптомное или малосимптомное течение, обычно без приступов острого панкреатита;
• иногда формирование панкреатических кальцификатов и кист;
• иногда сочетание с другими аутоиммунными заболеваниями;
• эффект от лечения глюкокортикостероидами (ГКС).
Предлагались и другие названия ХП с подобной клинической картиной: хронический воспалительный склероз ПЖ, склерозирующий панкреатит, панкреатит с сужением протока, склерозирующий панкреатохолангит. Однако в случае отсутствия доказательств аутоиммунного процесса эти формы должны рассматриваться как иные заболевания – даже в случаях эффективности стероидной терапии. Лишь недавно термин “аутоиммунный панкреатит” стал общепринятым, хотя очевидно, что он является гетерогенным заболеванием и может быть как первичным (самостоятельным), так и вторичным (в структуре синдрома Съегрена, воспалительных заболеваний кишечника – ВЗК) [5, 6]. В последние годы интерес к АП существенно возрос, поскольку появилась возможность выявления новых маркеров болезни, а также проведения биопсии ПЖ.
Эпидемиология
АП – редкое заболевание. Несмотря на то что за последние 10 лет количество сообщений о случаях АП в литературе растет, общее число пациентов остается незначительным. Распространенность и заболеваемость АП неизвестны, отдельные исследования указывают, что АП составляет примерно 5–6 % среди ХП у взрослых [7]. В США среди 254 больных с ХП у 27 на основании гистологических данных был установлен диагноз АП, что составило 11 % [8]. В Японии К. Okazaki и Т. Chiba [9] выявили АП у 21 из 451 пациента с ХП (4,6 %). Клинические и иммунологические признаки АП были обнаружены у 40 % больных идиопатическим ХП [10]. Заболевание встречается у лиц обоего пола, но у мужчин примерно вдвое чаще, чем у женщин. Оно возможно в любом возрасте, но большинство пациентов – лица старше 50 лет [11]. Описания у детей единичны и касаются в основном случаев вторичного поражения ПЖ на фоне системных заболеваний.
Этиология и патогенез
Причины АП неизвестны, но имеющиеся данные подтверждают аутоиммунный механизм его развития [12, 13]. Как и другие аутоиммунные поражения, АП может быть ассоциирован с ревматоидным артритом, ВЗК, синдромом Съегрена. Это сочетание указывает на вероятную общность иммунологических мишеней в некоторых органах.
Иммунологически АП характеризуется повышением уровня γ-глобулинов и особенно IgG4 в крови, а также присутствием антинуклеарных антител (АНА), антител к карбоангидразе II (АКА II) и лактоферрину (АЛ). Хотя повышение IgG4 связано с АП, эти неспецифичные и функционально
моновалентные антитела скорее являются вторичным ответом на неизвестный пока триггер воспалительного процесса. Аутоантитела (АНА, АКА II, АЛ) могут служить потенциальными иммунологическими серологическими маркерами АП [12, 14]. В аутоиммунный процесс при АП могут вовлекаться и другие органы: билиарный тракт, канальцы почек и легкие; все они содержат внутриклеточную цитоплазматическую карбоангидразу и лактоферрин [15]. В эксперименте на мышах сходное с АП поражение ПЖ было получено после тимэктомии и иммунизации животных лактоферри-
ном или карбоангидразой [14].
По сравнению с другими формами ХП у больных АП повышен уровень CD4+- и CD8+-Т-лимфоцитов, несущих HLA-DR [11]. Введение экспериментальным животным CD4+- Т-лимфоцитов, сенсибилизированных к амилазе, вызывало развитие модельного АП [13]. CD3+-T-лимфоциты
превышают В-лимфоциты в инфильтрате ПЖ, хотя и В-клетки, и плазмоциты, и лимфоидные фолликулы также могут присутствовать [16]. Адаптивный иммунный ответ при АП первоначально характеризуется преимущественной дифференцировкой Th1 c продукцией интерферона γ и фактора некроза опухоли α; позднее происходит повышение Th2-ответа, что обычно сопряжено с прогрессированием заболевания [9]. Не исключена генетическая предрасположенность к АП. S. Kawa и соавт. [17] обнаружили ассоциацию DRB1*0405–DQB1*0401 HLA гаплотипа с АП у японцев.
Примерно у половины больных АП развивается сахарный диабет, при этом могут быть обнаружены аутоантитела к островковым β-клеткам ПЖ, глютамилдекарбоксилазе и подобному тирозинфосфатазе белку [18]. При вторичном АП в структуре синдрома Съегрена возможны антитела к α-фодрину [19].
Морфология
ПЖ при АП диффузно увеличена и уплотнена, иногда изменения имеют более локальный характер. Морфологически для АП характерна перидуктальная лимфоплазмоцитарная инфильтрация [5]. Иногда обнаруживают перидуктальные эпителиоидно-клеточные гранулемы. Большинство лимфоцитов инфильтрата относится к CD8+- и CD4+-Т-лимфоцитам. Внутридольковые перегородки утолщаются за счет пролиферации миофибробластов и лимфоплазмоцитарной инфильтрации [20, 21].
При вовлечении других органов в желчном пузыре, желчных протоках, почках, легких и слюнных железах могут также выявляться участки воспаления с плотной лимфоплазмоцитарной инфильтрацией, сопровождающейся пролиферацией миофибробластов. Как в ПЖ, так и в других органах обнаруживают IgG4-позитивные плазматические клетки [22]. Возможны очаговые поражения желудка, двенадцатиперстной и толстой кишок [23].
Клиника
Клиническая картина АП может быть весьма вариабельной, но сильные боли в животе и острый панкреатит не очень характерны. Обычно беспокоят умеренные рецидивирующие боли, реже наблюдается небольшая холестатическая желтуха, связанная с сужением общего желчного протока в его панкреатической части. Среди 24 взрослых пациентов с АП, наблюдавшихся D. Finkelberg и соавт. [24], у 63 % отмечена небольшая желтуха и лишь у 35 % – боли в животе. В половине случаев развивается сахарный диабет. Важно отметить, что все симптомы ослаблялись на фоне лечения ГКС. Иногда клиническая манифестация АП уходит на второй план и характеризуется симптомами со стороны других органов, которые вовлекаются в патологический процесс. АП может ассоциироваться с ВЗК, чаще с неспецифическим язвенным колитом. Так, по данным G. Zamboni и соавт. [5], ВЗК были диагностированы у 9 (17 %) из 53 пациентов с АП. При АП могут развиваться стриктуры в панкреатической части холедоха в виде диффузного длинного сегмента сужения, что отличает его от ПСХ, при котором стриктуры желчных протоков прерывисты, ветвисты и напоминают бусы или четки [25]. Стриктуры холедоха при АП в отличие от ПСХ хорошо поддаются консервативной гормонотерапии.
Экстрапанкреатическая манифестация АП может вовлекать почки и легкие. В легких появляются очаговые или диффузные узелковые инфильтраты, иногда наблюдается аденопатия легких. При вовлечении почек развивается очаговое поражение в виде деструктивной лимфоплазмоцитарной инфильтрации с IgG4-позитивными плазмоцитами, возможна легкая почечная недостаточность [26, 27].
Диагностика
Диагностика АП базируется на сочетании клинических, иммунологических данных и результатов визуализирующих методов исследования. Классические изменения со стороны ПЖ при исследовании с помощью компьютерной томографии (КТ) характеризуются диффузным ее увеличением в форме сосиски с некоторым гомогенным умеренным уплотнением и появлением “ореола” вокруг. Дольковая структура обычно не прослеживается. Поражение перипанкреатической клетчатки минимально. В области головки могут выявляться более локальные очаги уплотнения или, напротив, размягчения ткани, что создает трудности при дифференциальном диагнозе с опухолями ПЖ. При длительном течении АП, как правило, определяются и изменения в области хвоста ПЖ. Возможно некоторое увеличение регионарных лимфоузлов. На фоне лечения ГКС указанные изменения уменьшаются уже через 1–2 недели и в дальнейшем могут полностью исчезнуть.
Изменения протоков, по данным РХПГ или магнитно-резонансной холангиопанкреатографии (МРХПГ), характеризуются лишь незначительным диффузным или сегментарным сужением холедоха, без дополнительных стриктур и разветвлений [7, 28]. Общий желчный проток и внепеченочные протоки могут быть несколько расширенными выше зоны сужения.На фоне применения ГКС эти изменения проходят [29].
Появление эндоскопических ультразвуковых исследований (ЭУЗИ) открыло новые возможности в диагностике патологии ПЖ, в частности детальной визуализации ее структуры, а также тонкоигольной прицельной биопсии и аспирации. Наиболее часто при АП ПЖ по данным ЭУЗИ диффузно увеличена, гипоэхогенна, очагово уплотнена, может выявляться расширение вирсунгова протока [30, 31]. Исследование биоптата ПЖ позволяет подтвердить диагноз АП гистологически [31].
Традиционное трансабдоминальное УЗИ может выявлять изменения ПЖ, однако они не имеют какого-либо специфического характера и не дают возможности дифференцировать АП от других форм панкреатита. Улучшение ультразвуковых данных на фоне лечения ГКС косвенно свидетельствуют в пользу АП [29].
Диагностические критерии
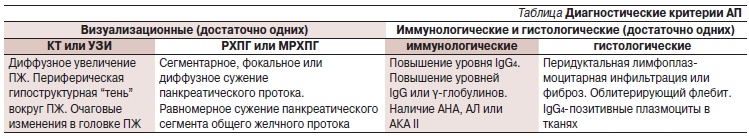
Диагностические критерии АП были предложены Японской панкреатической ассоциацией с целью дифференцировать АП от других форм ХП [32]. Критерии базируются на комбинации данных визуализирующих, лабораторных исследований и биопсии (см. таблицу). Предложены и другие мето-
ды оценки, в частности сочетание клинических, визуализационных, лабораторных и гистологических данных, а также учет эффективности гормонотерапии [33].
Таблица. Диагностические критерии АП.
Гистологическая диагностика не считается обязательной для постановки диагноза АП, поскольку лимфоплазмоцитарная инфильтрация и фиброз ПЖ могут быть обнаружены при других формах ХП, а процедура получения биоптата ПЖ – весьма сложная и инвазивная. Лапароскопия, биопсия и морфологическое исследо-вание показаны лишь при подозрении на опухоль ПЖ.
При клинической картине панкреатита диагностический алгоритм начинается с методов визуализации. При обнаружении типичной картины проводятся иммунологические тесты, прежде всего оценка уровня IgG4. Если содержание IgG4 повышено, то в сочетании с данными визуализационной диагностики можно говорить о вероятном АП, и этого достаточно для назначения гормональной терапии. Если при этом выявлены аутоантитела, диагноз считается установленным. Положительный эффект гормонотерапии является важным аргументом для окончательного подтверждения диагноза АП.
Лечение
Если диагноз АП установлен на основании визуализационных и иммунологических методов, показано назначение ГКС, и обычно с этой целью используют преднизолон в дозе 1 мг/кг в день (взрослым 40 мг в день). Эта доза назначается на неделю, затем ее снижают по 5 мг в неделю, оставляя поддерживающую дозу на несколько месяцев. Обычно на фоне терапии ГКС наблюдается быстрое клиническое и лабораторное улучшение [34–36]. Выявляемые на УЗИ и КТ изменения ПЖ уменьшаются и исчезают, снижается уровень IgG4. В том случае если на фоне гормонотерапии улучшения не наблюдается, диагноз АП следует считать ошибочным и необходим поиск других причин поражения ПЖ.
Лечение больных с выявленной по данным теста на эластазу-1 в кале внешнесекреторной недостаточностью ПЖ на фоне ХП должно включать панкреатические ферментные препараты. Выбор препарата должен основываться на следующих критериях:
1. Высокое содержание липазы в препарате. Липаза является самым уязвимым ферментом, поскольку разрушается химотрипсином и работает только в проксимальных отделах тонкой кишки. Расчет дозы фермента производится по липазе, для стартовой терапии обычно достаточно 1000 ЕД на 1 кг массы в сутки, при недостаточной эффективности дозу повышают в 2 раза, затем подбирают индивидуально. Критерием адекватности дозы являются исчезновение диспепсических расстройств и достаточные весовые прибавки. Суточную дозу делят на все приемы пищи; обычно в основные приемы пищи доза в 2 раза выше, чем при перекусах.
2. Соотношение колипазы к липазе выше 1. У Креона это соотношение наибольшее и составляет 1,9.
3. Наличие оболочки, защищающей препарат от воздействия желудочного сока. Основные ферменты – липаза и трипсин, разрушаются при кислых значениях рН: липаза – при рН ниже 4, трипсин – ниже 3, для эффективного действия липазы необходимо поддерживать рН выше
4 в желудке в течение 60 минут после приема препарата, в двенадцатиперстной кишке – в течение 90 минут. Наилучшим способом защиты являются кислотоустойчивые микросферы, которые растворяются при рН выше 5,5, т. е. в двенадцатиперстной кишке.
4. Равномерное поступление препарата в двенадцатиперстную кишку вместе с пищей. Это обеспечивает активное участие ферментов в процессе полостного пищеварения с самого начала поступления химуса в кишечник. Привратник не пропускает крупные частицы (более 1,4 мм),
поэтому оптимальной формой для ферментных препаратов являются минимикросферы диметром менее 1,4 мм. Минимикросферы Креона имеют самый малый диаметр – 0,8–1,0 мм, что позволяет препарату беспрепятственно проникать в кишечник и с самого начала активно участвовать в процессе пищеварения.
Больной должен получать достаточное по количеству белка и калорий питание, ограничение касается в основном жирной пищи и экстрактивных веществ. Необходимо обеспечивать достаточное поступление витаминов и микроэлементов. Терапия обострения АП предполагает функциональную разгрузку ПЖ, прежде всего уменьшение стимулирующих влияний секретина и холецистокинина.
Последний стимулируется пептидами, поэтому назначение ферментных препаратов, расщепляющих белки в полости тонкой кишки, по принципу обратной связи способствует уменьшению стимулирующих влияний на ПЖ. На фоне обострения АП оптимальны панкреатические ферменты с более высоким содержанием протеиназ. Специальных препаратов для этой цели не существует, поэтому на практике используют те же ферментные средства, что и для замести тельной терапии, но в более высоких дозах. Оптимальным препаратом является Креон®, который благодаря микросферической форме обеспечивает максимальное поступление ферментов в двенадцатиперстную кишку вместе с пищей.
Длительность ферментотерапии может варьироваться. При незначительных степенях внешнесекреторной недостаточности ПЖ на фоне обострения возможно последующее частичное восстановление ее функции, поэтому курс лечения может быть ограниченным 2–3 месяцами с последующей отменой и повторной оценкой экзокринной функции. Функциональный покой на фоне ферментотерапии может способствовать улучшению морфологического и функционального состояния ПЖ, вопрос о необходимости повторных курсов или постоянной заместительной терапии решается индивидуально.
При стойком снижении уровня эластазы-1 в кале показана постоянная заместительная ферментотерапия.
Результаты собственных наблюдений
Из 57 наблюдавшихся нами детей с установленным на основании клинических, биохимических и визуализирующих (ЭУЗИ, КТ) методов диагнозом ХП аутоиммунная этиология заболевания была установлена у 4. При постановке диагноза АП мы пользовались критериями, приведенными выше. Под нашим наблюдением находились два ребенка с первичным АП и еще два – с вторичным АП, у которых заболевание развилось на фоне болезни Крона, у одного из них – с синдромом Съегрена, проявившимся атрофическим кератитом, сухостью слизистой оболочки рта. Дети с болезнью Крона и вторичным АП не предъявляли типичных для панкреатита жалоб на боли в животе, но при углубленном обследовании (УЗИ, ЭУЗИ) у них были выявлены признаки поражения ПЖ (увеличение размеров, признаки фиброза, расширение вирсунгова протока у одного пациента). Заболевание не сопровождалось признаками ПСХ. Иммунологическое исследование обнаружило у обоих пациентов повышение уровня IgG4, АНА, антитела к двуспиральной ДНК, внутриорганные антитела к ПЖ. Амилаза крови оставалась нормальной или была незначительно повышенной. Уровень эластазы-1 в кале был снижен, особенно у пациента с синдромом Съегрена (54 мкг/г).
Первичный АП, установленный нами у двух детей, протекал как изолированное самостоятельное заболевание. Обе наблюдавшиеся нами больные были девочками дошкольного возраста. Заболевание в обеих случаях протекало ярко и манифестно. Пациентки поступили с жалобами на сильные боли в области пупка, многократную рвоту с примесью желчи, разжиженный стул. Уровень амилазы был повышен в 10 и более раз. По данным УЗИ отмечено увеличение размеров ПЖ с неравномерным повышением эхогенности. Для подтверждения диагноза в обоих случаях была проведена КТ; в обоих случаях выявлены признаки хронического воспаления и фиброза в ПЖ с расширением вирсунгова протока. Уровень эластазы-1 в кале был снижен. В крови обеих больных обнаружены повышенный уровень IgG4, а также антитела к лактоферрину и антиорганные антитела к ткани ПЖ, у одной из них – также к островковым β-клеткам. Заболевание характеризовалось рецидивирующим течением с типичными болевыми приступами, повышением уровня амилазы и диспепсическими расстройствами. Обострения обычно провоцировались нарушением диеты или вирусной инфекцией. В обоих случаях положительный эффект был достигнут после назначения преднизолона.
Клинический пример
Больная Даша Т. 4 лет поступила в ДГКБ № 5 им. Н.Ф. Филатова Санкт-Петербурга 06.01.2007 с жалобами на боли в животе, преимущественно в области пупка, многократную рвоту с примесью желчи; стул оформленный. Больна в течение суток. Данный приступ третий по счету в течение года (первый – в апреле 2006-го, второй – в июне 2006 г.). Оба предыдущих приступа протекали аналогично: боли в животе, повторная рвота с примесью желчи, нормальный стул, нормальная температура. Во время предшествующих приступов отмечено повышение амилазы крови до 316 и 1197 ЕД/л (при норме до 125 ЕД/л), размеры и структура ПЖ по данным УЗИ не были изменены. После второго приступа был установлен диагноз “рецидивирующий панкреатит”, девочка находилась на диете. Ранний анамнез без особенностей. Росла и развивалась соответственно возрасту. Отмечались аллергические реакции на цитрусовые и пенициллин в виде сыпи. Привита по возрасту. Случаев панкреатита, сахарного диабета среди родственников не имелось.
Состояние при поступлении тяжелое. Температура нормальная. Умеренно выражены признаки эксикоза, вялая, сознание ясное. Кожа бледная, чистая, мраморность в дистальных отделах конечностей. Слизистые оболочки суховаты, розовые, язык обложен белым налетом. Тоны сердца ритмичные, звучные, тахикардия 123 уд./мин, артериальное давление – 120/60 мм рт. ст. Дыхание в легких везикулярное, хрипов нет. Живот мягкий, умеренно болезненный в области пупка, реагирует на глубокую пальпацию в зоне проекции поджелудочной железы. Печень и селезенка не увеличены. Стул после клизмы оформленный. Мочеиспускание свободное, безболезненное.
В клиническом анализе крови: гемоглобин – 133 г/л, эритроциты – 4,7 × 1012/л, лейкоциты – 17,7 × 109/л, палочкоядерные – 1, сегментоядерные – 89, эозинофилы – 0, базофилы – 0, лимфоциты – 5, моноциты – 5, СОЭ – 20 мм/ч. В биохимическом анализе крови: амилаза – 1565 ЕД/л, глюкоза – 5,6 ммоль/л, АЛТ – 18,1 ЕД/л, АСТ – 32,2 ЕД/л, мочевина – 5,4 ммоль/л, общий белок – 76,4 г/л, ЛДГ – 578 ЕД/л, ГГТП – 11 ЕД/л, билирубин общий – 16,4 ммоль/л, прямой – 1,1 ммоль/л, холестерин – 3,3 ммоль/л, триглицериды – 0,3 ммоль/л, β-липопротеиды – 358 ЕД. Анализы мочи и кала в норме, посев кала – отрицательный. Дневные колебания глюкозы в крови (17.01.2007): натощак – 3,9 ммоль/л, через час после еды – 8,8 ммоль/л, через 2 часа – 5,4 ммоль/л.
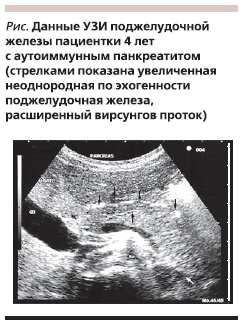
УЗИ брюшной полости (см. рисунок): ПЖ увеличена в размерах (головка – 1,6, тело – 1,6, хвост – 2,3 см), структура однородная, эхогенность несколько повышена, капсула четко контурируется. Вирсунгов проток расширен, просвет его – до 1,2 мм, стенки уплотнены, прослеживаются боковые протоки. Желчный пузырь умеренно увеличен (7,0 × 2,4 см), холедох – 0,2 см, стенки ровные, не утолщены, внутриполостных сигналов нет. Печень, почки, селезенка – без патологии. Иммунологическое исследование крови (19.01.2007): IgG1 – 6,0 мг/мл (норма – 2,3–6,4 мг/мл), IgG2 – 4,0 мг/мл (норма – 0,3–4,5 мг/мл), IgG3 – 1,2 мг/мл (норма – 0,1–1,1 мг/мл), IgG4 –1,8 мг/мл (0,1–0,8 мг/мл).
ANA – 1 : 120, антитела к островковым клеткам поджелудочной железы – 1 : 10 (норма меньше < 1 : 5). Антиорганные антитела к поджелудочной железе – 2,5 (норма < 1,1).
С учетом повторного характера приступов, клинических и ультразвуковых данных, 15-кратного увеличения уровня амилазы в крови, обнаружения аутоантител к поджелудочной железе, ANA и повышенного уровня IgG4 в крови был установлен диагноз: аутоиммунный панкреатит, угрожаемый по сахарному диабету.
После коррекции водно-электролитных нарушений и установления генеза панкреатита была начата терапия преднизолоном в дозе 1 мг/кг/сут. Для купирования приступа назначались также омепразол по 1 мг/кг/сут и Креон® по 30000 МЕ липазы в сутки. На фоне лечения произошла быстрая нормализация уровня амилазы в крови, при повторном УЗИ – через 2 недели: ПЖ нормальных размеров (головка – 1,5, тело – 0,9, хвост – 1,0 см), вирсунгов проток не визуализируется. Дневные колебания глюкозы в крови: натощак – 4,4 ммоль/л, через час после еды – 5,9 ммоль/л, через 2 часа –5,2 ммоль/л.
После постепенного снижения дозы преднизолона и его отмены через 6 месяцев состояние ребенка оставалось удовлетворительным, но спустя 2 месяца она вновь была госпитализирована с приступом панкреатита. По данным УЗИ ПЖ увеличена в размерах, эхогенность неравномерная, с участками фиброза, незначительное расширение вирсунгова протока. Амилаза крови – 1820 ЕД/л. Уровень эластазы-1 в кале – 92 ЕД/г. Произведена МРХПГ, изменений в желчных протоках и протоках ПЖ не выявлено.
Вновь начата терапия преднизолоном в дозе 1 мг/кг/сут. С учетом снижения уровня эластазы-1 в кале был назначен Креон® в дозе 20000 МЕ липазы в сутки. Состояние девочки улучшилось, уменьшились размеры ПЖ, но уровень амилазы оставался немного повышенным (130–150 ЕД/л). С учетом рецидивирующего характера заболевания, обострения после предыдущей отмены преднизолона на фоне снижения дозы во время повторного курса преднизолона был назначен меркаптопурин в дозе 1 мг/кг/сут.
Девочка наблюдается нами до настоящего времени, в течение 2 лет на фоне приема меркаптопурина и Креона обострений панкреатита не наблюдалось, уровень амилазы нормальный, размеры ПЖ по данным УЗИ в пределах нормы, вирсунгов проток не расширен. Эластаза-1 в кале – 320 мг/г.
Данный случай подтверждает возможность развития АП в раннем детском возрасте как первичного заболевания, без какой-либо фоновой системной патологии. Обнаружение аутоантител и повышенный уровень IgG4 могут служить маркерами аутоиммунного генеза ХП у детей. Особенностью первичного АП в детском возрасте, по нашему мнению, является его манифестное течение с тяжелыми обострениями и формированием не только экзокринной недостаточности, но и сахарного диабета. Терапия преднизолоном при АП у детей эффективна и приводит к быстрому обратному развитию изменений ПЖ, а также устранению симптомов заболевания. Однако эта терапия не предотвращает возможность рецидивов. Назначение тиопуринов (меркаптопурина) при тяжелом рецидивирующем АП, как показал этот случай, позволяет длительно поддерживать ремиссию заболевания и способствует не только исчезновению симптомов, но и улучшению морфологической структуры и функции ПЖ. Ферментотерапия с использованием микросферических панкреатических ферментов (Креон®) является неотъемлемой частью комплексного лечения обострений АП, а также необходимым элементом устранения функциональной недостаточности ПЖ.



{kind=link}